
2024-05-08 来源 : 胖猫的生命医学札记

引言:过去几十年中,FDA为鼓励罕见病和严重疾病创新药的开发制定了一系列的特殊审评通道,包括:孤儿药、快速通道、加速审批、优先审评和突破性疗法认定。
常常听到,常常忘记,但是还是“傻傻分不清楚”,这篇就系统整理,哪天记不起来了,就翻出来看看,记得点赞收藏,关注星标不迷路啊。
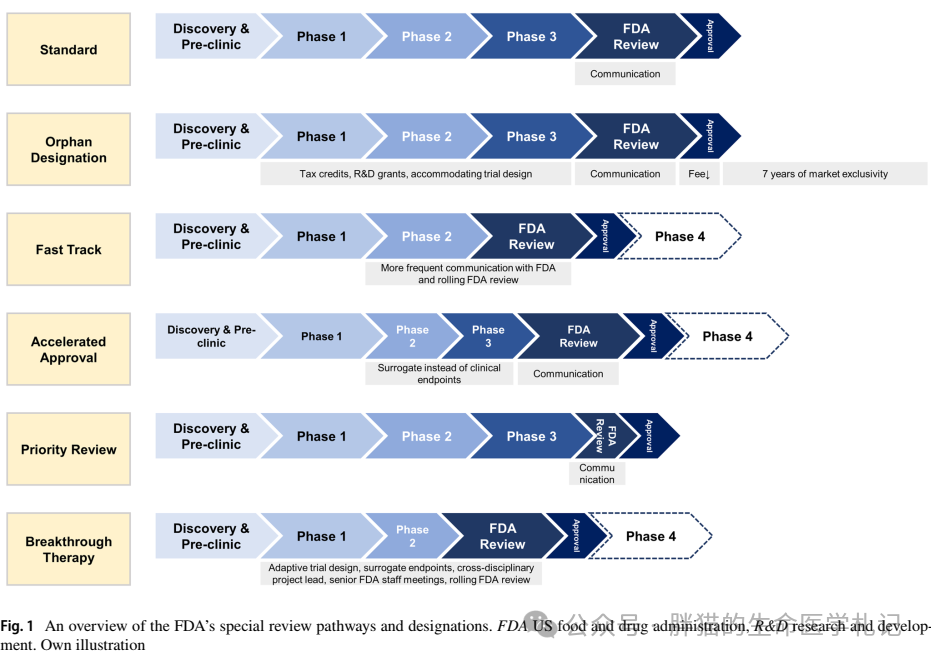
图注:FDA“特殊审评”政策和标准流程的对比示意图
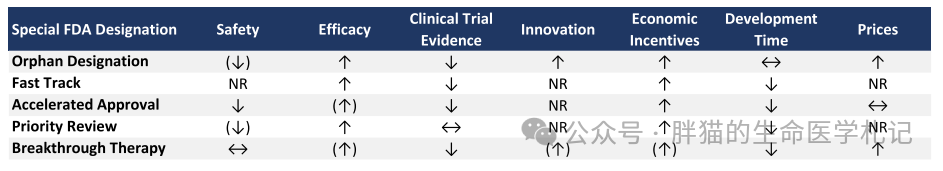
由此带来的财政激励、开发时间缩短、更高临床试验成功率及更高的价格使得药企投资这类药物动力十足,但特殊通道的使用(或潜在滥用)在科学、医疗保健政策和公众中引发了广泛争论。
特殊通道可能带来未识别的不良事件和上市后安全问题(如撤回或警告),因为支持特殊批准的临床试验经常使用小型、非随机、开放标签的设计,且用于监测药物不良事件的上市后临床(IV期临床),经常被延迟甚至不启动。

图注:2003-2022年,通过FDA“特殊通道”政策审评的药物。
下文对着几种“特殊通道”进行简述~
孤儿药认定
Orphan drug designation
罕见病患者人数少,无法通过药物销售回收研发成本,这导致药企没有动力开发罕见病药物,由此也带来大量未被满足的临床需求——到20 世纪 80 年代,美国有 20-2500 万公民患有5,000 种罕见疾病。
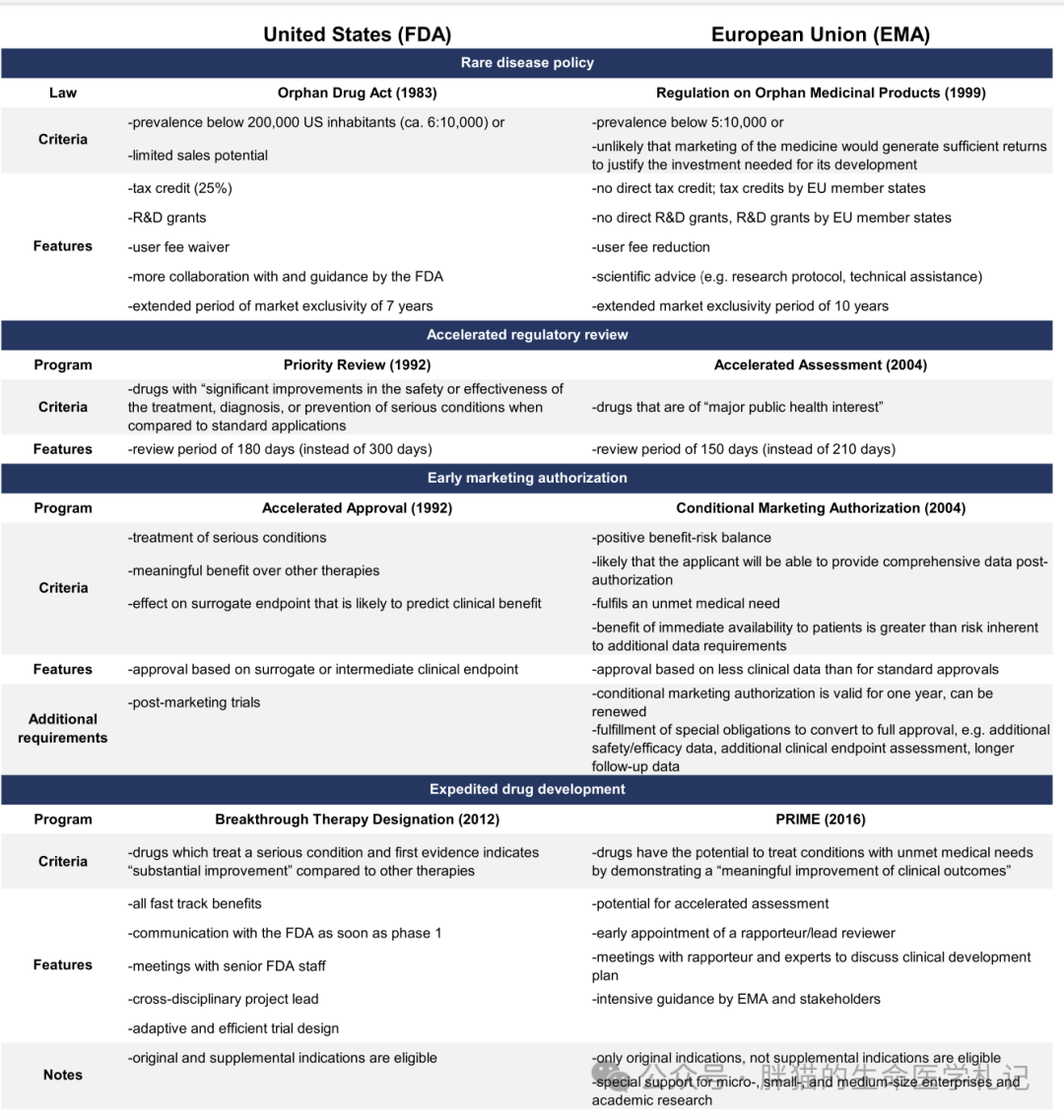
鉴于此,美国国会于 1983 年推出《孤儿药法案》(ODA),提出一系列推动罕见病药物研发的政策,如,税收抵免(临床研发成本的25%),为孤儿药项目提供研究资助,免除NDA/BLA费用(~ 300 万美元),上市后7年市场独占期等。此外,临床试验期间药企还可与FDA 对临床试验的设计进行紧密沟通,缩短 IND 和 NDA 的时间。

图注:FDA历年孤儿药的认定和获批情况
ODA政策取得了巨大成功。从 1983 年到2021 年底,FDA 授予了 6,143 种药物孤儿药资格,其中1,033种获批准。到 2002 年,这些药共为约 1100 万患者提供了新的治疗方法。尽管如此,孤儿药仅覆盖了 15% 的罕见病,且只有 5% 的药物获得FDA 批准。
孤儿药的审批途径也面临着挑战。基于1999 年至 2008 年间214 种孤儿药数据分析发现,69% 的药物在批准后,更新了标签中安全相关内容,其中 15% 是严重的安全事件,如撤回、警告或暂停。另一研究甚至观察到,EMA 批准的孤儿药有87%出现了严重的安全事件。这些安全问题部分可归因于非随机开放标签单臂试验的设计,试验规模只有非孤儿药临床试验的一半,这种小型、不稳健的试验具有较高的偏倚风险,可能夸大治疗结果。
孤儿药的获益和成本也被广泛研究。以中位数增量质量调整生命年 (QALY) 来衡量的话,孤儿药相对于非孤儿药获得了更高的健康收益(0.25 vs. 0.05 QALY)。对FDA 批准的 455 种肿瘤药物研究发现,孤儿药相对于非孤儿药将无进展生存期 (PFS) 分别延长了3.3 个月和 2.8 个月,因此与非孤儿药相比,孤儿药对具有更高的治疗价值。
从患者负担看,美国孤儿药的月均价格高于非孤儿抗肿瘤药(33,070 $ vs. 14,508 $),孤儿药成本显著高于非孤儿药。
从开发成本看,基于 2003 年至2011 年正在开发的 5820 个药物分析发现,孤儿药从I期到NDA机会高于平均水平(32.9% vs. 10.4%)。另一项对2006 年至 2015 年间的 7455 个开发项目的研究发现,罕见疾病药物的总体成功率为 25.3%,而平均成功率为9.6%。另外,由于成功率较高且入组患者较少,与非孤儿药相比,孤儿药将新药推向市场的临床开发成本低 29%。然而,由于罕见病患者招募困难且缓慢,孤儿药的临床开发时间没有显着差异。但是对于超级罕见病(患病率 < 6,600),临床试验比进行罕见或常见疾病的试验复杂得多,因为申办者很难找到足够数量的患者、有能力的研究人员和可以实施治疗的医疗中心。
孤儿药政策也得到制药公司和投资者的认可。根据 2005 年至2020 年期间的 311 起并购事件分析,投资于孤儿药开发公司的年回报率更高(46% vs. 12%)。此外,对同时用于治疗罕见病和常见病的孤儿药——“部分孤儿药”也有大量的争议,药企首先谋求药物的孤儿适应症,然后扩展到非孤儿适应症(“孤儿优先策略”)。这些“部分孤儿药”比孤儿药更有吸引力,常会成为“重磅炸弹”, 2019 年,十大最畅销药物中有7 个对“部分孤儿药”,最终受益高于孤儿药溢价。
快速通道
Fast track
20 世纪 80 年代,HIV肆虐全球,一度亟需新的抗病毒药物来应对这种新出现的病毒。鉴于这一新出现威胁,美国于1988年推出快速审评通道计划,以“促进治疗严重疾病和满足未满足的医疗需求的药物的开发和加快审查”。
严重疾病的定义是一个相对主观的问题,但可以根据生存率、生活质量或疾病进展特征来评估。根据这个定义,严重疾病包括了艾滋病、痴呆、癌症、心力衰竭、癫痫、抑郁症和糖尿病。在这种情况下,FDA 将严重疾病药物解释为治疗无可用药物的疾病或比现有疗法更好的新药。药物的更优特点可以通过更好的疗效、更少的副作用、早期诊断带来更好的临床结果、更好和更长的依从性或满足未来的公共卫生需求来证明。
在快速通道流程下,制药公司成I期试验后与FDA 讨论II期试验设计,如果成功,II期试验(而非III期)将足以证明药物的安全性和有效性。根据该计划,FDA 可以持续审查临床试验产生的证据(滚动审查),不过 FDA 在出现不确定的副作用、毒性或治疗结果的情况下会要求进行上市后试验。

图注:FDA历年“Fast Track”审评药物
快速通道不仅向患者、医生和保险公司,也向投资者发出了积极信号。从 2003 年到2022 年,平均有 33% 的新药通过快速通道进行审评。在 1999 年至2012 年间批准的 135 种药物中,获得快速通道指定的药物提供了 0.254 增量QALY 的中位效益,而未指定药物则提供了 0.014 QALY 的中位获益。
加速审批
Accelerated approval
1992年,美国通过引入加速审批计划进一步加快药物研发。与快速通道类似,特定有价值的药物可以获得加速批准,从而满足未满足的临床需求。
加速批准使 FDA根据替代指标而非临床终点来判断药物的疗效。衡量药物对患者生存的影响可能需要较长的试验时间和随访数据(特别是对于 5 年和10 年生存率较高的癌症类型,例如前列腺癌或乳腺癌)。相比之下,替代终点(例如肿瘤缩小或进展)可以更快地观察到。因此,加速审批计划通过缩短临床试验持续时间并启用较少入组患者的试验设计来测量替代终点,从而加快了药物开发。
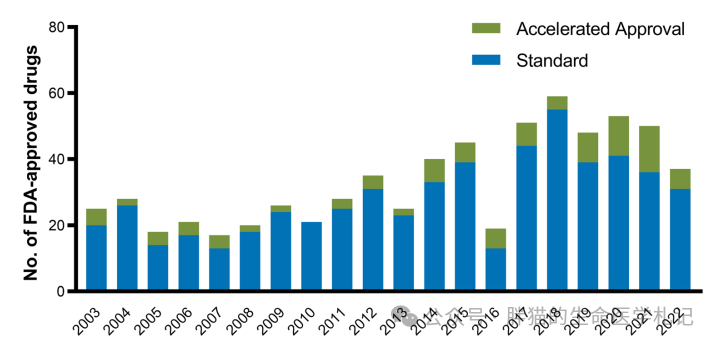
图注:FDA历年加速审评药物
2003 年到2022 年,平均有 15% 的药物获得加速批准,对 1999 年至2012 年间批准的 135 种适应症的研究,加速批准药物的 QALY 增量中位数较高(0.370 VS 0.031)。据观察,加速批准的抗癌药物比 FDA 标准批准的药物上市时间快3.9 年。对FDA 批准的 188 种癌症适应症的数据研究发现,使用 PFS 和肿瘤缓解率作为替代终点可分别将临床试验时间缩短了 11 个月和19 个月。
然而,较短和较小的试验对正确评估药物的风险和益处提出了挑战,加速批准的药物会导致更多未被识别的不良事件和上市后安全性修改,因此需要进行长期随访的大型试验,以确定药物的副作用及药物相关风险。然而,IV期试验常被推迟或未启动。更无语的是,IV期试验甚至会发现药物无效或有害,导致退市。
尽管加速药品的价格幅度与标准审批相比没有差异,但其不确定的证据基础给保险公司和支付方带来了重大挑战。如果没有临床结果数据,保险公司和支付方无法充分衡量药物相对于标准治疗的增量价值,从而难以决定药物的价格、报销和承保范围。
优先审查
Priority review
在通过加速审批途径的同年,1992年美国也引入了优先审查。FDA 根据 NDA 药物的疗效分配资源,分三个等级(A、B或 C)。根据新的政策,这三个类别被简化为两类:优先审查或标准审查。

2021 年 50 种新药中有34 种 (68%) 受益于优先审评。平均55% 的新药受益于优先审评。优先审查的适应症显示出比标准审查批准的适应症为患者带来更大的中位健康增益(0.175 vs. 0.007 QALY)。然而,较短的审查时间也与更多的批准后安全修订有关。优先审评下批准的药物在 FDA 批准后进行标签修订的可能性是标准审评下批准的药物的两倍。
突破性疗法认定
Breakthrough therapy designation
2012 年,美国国会引入了新的 FDA 审查途径:突破性疗法。FDA认为生物技术的发展,如下一代测序、基因疗法或细胞疗法,以及药物开发方式的进步(如精准医学或 RWE),需要在监管审批过程中得到反映。根据这一论点,创新药物(如靶向疗法或基因疗法)的疗效已经可以在II期试验中观察到,因此大规模随机对照试验(RCT)显得没那么必要。
该认定提供与 FDA 高级人员更早(I期)和更频繁的会议,以指导有效的药物开发和监管审查流程。此外,突破性疗法指定的药物可以获得快速通道计划的所有好处。

图注:FDA历年突破性疗法认定。a,突破性疗法审评情况;b,突破性疗法审批上市情况。
截至2022 年底,FDA 共收到 1,289 项突破性治疗请求,其中506 项(39%)获得批准,125 种突破性新药获NDA。研究发现,基于开放标签盲法的非随机单组研究,突破性药物更容易获得批准,与非突破性癌症药物相比,突破性药物的临床开发时间要快2-3 年。从患者负担来看,突破性药物的月治疗费用高出约 16,000 美元($38,971 vs. $22,591 )。
对 2012 年至2022 年 FDA 批准的 355 种突破性和非突破性肿瘤药物进行了比较发现,突破性适应症与非突破性适应症相比,死亡可能性较低(风险比:0.69 vs. 0.74),并且中位总生存期显着改善(4.8 VS 3.2 个月)。
另外,患者和医生对“突破”一词具有非常积极的反映。“突破”一词意味着这些药物是重大的科学颠覆,可能导致误导性的、毫无根据的乐观情绪。与仅事实描述相比,“突破”一词使更高比例的患者相信该药物有效(11% vs. 25%)。当在具有相同安全性、疗效、证据和成本的两种药物之间进行选择时,94% 的医生选择开出具有突破性称号的药物。
结语
Conclusion
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。
































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)