
2023-01-18 来源 : 英勇向前 ,作者徐应永

以FDA为代表的监管部门对抗肿瘤药物开发的诸多“新”要求,包括日益趋严的加速批准、探索推荐治疗剂量、伴随诊断的开发等,势必革新未来新药的开发模式。对以抗肿瘤药物开发为主的制药企业,特别是小型生物技术公司,可能会产生较大影响,务必未雨绸缪。
监管“新”要求
抗肿瘤药物的研发时间长、风险高、费用大。新药从进入临床研究研究,直至获批大约需要5-10年,而成功率只有约5-6%,单个新药的研发成本可以超过20亿美元。
为了提高研发生产率,有学者提出Chorus模型如下,即通过早期研究尽可能多地获得有效信息,避免低成功率的、昂贵的临床II/III期研究,节约的成本再投资早期研发,直到确认较高的成功性(PTS)后才进入后期开发。
在此模型直接或间接指导下,过去10年里,抗肿瘤新药的开发已经很少用到传统临床开发路径,转而通过合并的临床I/II期研究同步探索推荐的II期剂量(RP2D)、联合治疗、甚至获得监管部门的加速批准,与此同时进行临床三期确证性研究。
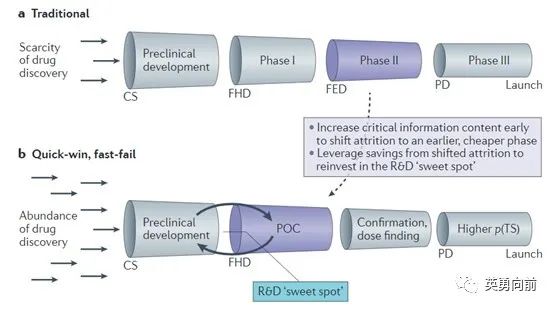
从包括监管在内的社会层面来说,具有高度医学需求的晚期肿瘤患者亟需有效的治疗手段,因此对于新药的需求是非常迫切的。基于此,FDA根据法律制定加速审批路径,包括快速通道、突破性疗法、加速批准和优先审评。这一措施大大加快了肿瘤患者获得新药的速度,为患者提供了新的希望,对整个社会也发挥了积极作用。
FDA的加速批准(AA)始于1992年,基于可预测临床获益的替代研究终点,针对严重或致命性疾病,给予提前批准。通常申办方将被要求进行三期确证性临床研究。
最初的AA用于治疗HIV药物的批准,血清mRNA水平作为替代终点预测HIV进展或死亡(临床获益)。过去10年里,85%的AA(172个适应症)为抗肿瘤药物,其中50%在确证性研究中被证明其临床获益,12%被撤回,38%仍在确证性研究中。中位至确证临床获益(常规批准)的时间为3.1年(0.5-17.6),中位至撤回时间为3.8年(1.3-12.5)。

抗肿瘤药物最常用的替代终点是持久的ORR,反应肿瘤体积缩小的状态,可以明确反应药物确实在发挥直接抗肿瘤作用。另一方面,ORR并不能完全等同于OS或生存质量的提高,这在PI3Ki的多个案例中均有报道。总体来讲,ORR只是体现了药物治疗疾病,而OS才是药物对患者的作用。
既要允许快速上市路径,又要避免不恰当的审批。特别是AA获批后到确证性研究的这段时间,假设确证性研究失败,这段时间越长则患者和社会付出的代价越大。基于此,FDA官员和学者在NEJM发表数篇短文,从如何批(on ramp)和如何撤(off ramp)这两方面。
On ramp:仅通过单臂研究想获得AA在将来可能会比较困难。无论如何分析,都很难把ORR等同于临床获益,而单臂研究又很难评估生存数据。
因此,FDA官员在文章中提出两种建议:
1)直接开展三期确证性研究,增加以ORR和DOR为终点的中期分析,该结果支持FDA的AA批准,而正式批准将基于最终结果。
2)同时开展上述两项研究(单臂和RCT),而RCT的中期ORR分析支持单臂研究的AA批准。该要求增加FDA批准的信心,同时缩短“无效上市”(从AA到因确证研究失败最终撤市)的时间。
Off ramp:撤回的关键问题在时间和方式。当确证性研究失败后,FDA建议后如果申办方不主动撤回,则需要通过听证会形式处理。唯一的案例是Avastin治疗转移性乳腺癌,这一听证过程费时16个月。
因此,为减少潜在风险,有学者建议规定申办方需要承诺在获得AA后5年之内完成确证性研究,或者在预计确证性研究完成后1年仍未完成该研究的,加速批准将自动失效,除非FDA认为确实为特例。

加速批准需要从三期确证性研究中获得早期数据以支持FDA的决策,因此关于三期研究设计,申办方应考虑如何在双盲设计时取得中期数据又不破坏研究完整性,同时也应考虑是否有足够资源,及时开展研究。
接下来学习第二个问题。
对抗肿瘤药物推荐II期剂量(RP2D)的探索,其基本逻辑仍在化疗时代。即,剂量越高、毒性越大、疗效越好,找到最大可耐受剂量(MTD)可以确定RP2D,也就是可以耐受的疗效最好的剂量水平。
化疗药物循此逻辑发明了自体干细胞移植,然而在小分子、抗体类、甚至细胞治疗广泛开发的今天,该逻辑存在很多现实问题。
首先,大分子抗体在临床一期方法学下,无论rule或model based,都很少达到MTD,指导MTD的DLT的定义也存在变异性。抗体类药物的PK相对复杂,抗体自身的代谢、晚期肿瘤患者情况、肿瘤负荷、抗药抗体(ADA)对药物剂量-效应关系均存在影响。并且临床前研究数据很难推导至人体研究,治疗窗和脱靶相应都不是很清楚。
小分子靶向药也可以细分为:高度选择性(sotorasib)、单激酶家族(erdafitinib)、多激酶抑制剂(lenvatinib),不同类型靶向药的治疗窗、脱靶效应均不尽相同。
其次,DLT的随访时间有限,大多在21-42天,过长的观察时间在临床上很难执行。然而,超过50%的严重毒性反应发生在DLT观察期以外。
无论是按照3+3、rolling six、快速滴定法,还是BION、mTPI等统计的方法学,剂量爬坡的逻辑是至关重要的。以MTD的方式寻找唯一的RP2D可能会导致后续一系列问题。例如下表格中列出的已进行过正式剂量调整的药物。
2021年5月,FDA加速批准Sotorasib治疗NSCLC的剂量为960mg每天(一次口服8片药物),但也是第一次FDA要求Amgen探索240mg剂量组,给出的5点理由可以参考,1. 稳态状态下的给药剂量(180-960mg)与药物暴露无相关性,2. 缺少明确的量效关系,3. 低级别剂量可能降低胃肠道反应,4. 临床前数据认为最小有效剂量为30-240mg,5. 说明书要求患者每次口服8片药物。

因此,FDA在2021年推出“Optimus 项目”,旨在研究更好的肿瘤产品剂量优化策略。
尽管没有透露更多细节,但确定剂量是的全面考虑是必要的,包括:1. 在确定最佳剂量时考虑所有信息;2. 尽早充分表征暴露/反应关系;3. 考虑超出剂量限度的毒性和首个治疗周期之后的信息;4. 在初始剂量递增后进行多剂量扩展队列 或随机。
近来,FDA针对新产品的开发,已纳入Optimus 项目进行指导,详细的方法学可能要根据具体案例来定。介绍一篇ESMO下属的MDICT给出了非常具体的建议,尽管并非官方意见,但非常值得仔细阅读和学习。

总结几点:
1.对一期研究的常用名称进行调整,例如:RP2D改为RD(recommend dosage)、MTD改为RDR(recommended dosage range)、DLT改为TLT(treatment limiting toxicity)。MTD与“Optimus 项目”不再相符,而RDR表示某个剂量区间,更为合理;RP2D并非只是II期剂量,改为RD更合理。
2.对于剂量探索的建议如下。简单来说:剂量确定要科学、方案设计要灵活。

本文给出了5种剂量探索的研究设计。
1, 研究分为剂量递增部分(dose escalation)和剂量区间研究(dosage ranging study)。递增不可以只看MTD确定单一RP2D,而是应综合考虑耐受性、MTD、疗效、PD/PK等数据综合决定,选择至少2个剂量进入区间研究,通过设计良好的随机对照研究,选择最佳给药方式(recommended dosage,RD)。
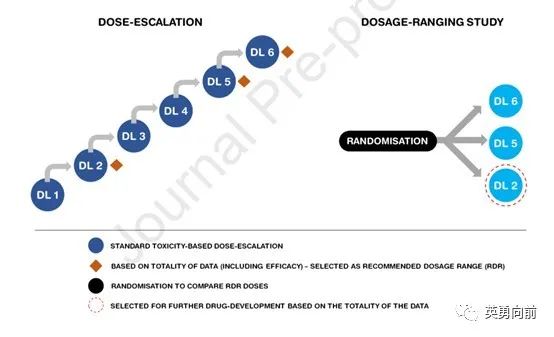
2, 基本逻辑时相似,相比第一种模式,可以在剂量递增阶段增加患者人数,更多患者数据将增加对推荐剂量区间(RDR)选择的准确性。
例如,当进入DL3同时,因观察到DL2伴肿瘤缩小,因此可额外增加DL2患者数,待DL6确定为MTD时,选择DL2和DL4进入剂量随机研究。

3, 把随机直接纳入剂量递增阶段,提高研究效率。例如,观察到DL2有疗效,则直接随机DL3和DL2,同理,DL4和DL2继续随机,最终确定DL2和DL4为RDR剂量。
随机后可以增加研究的科学性,进一步提高RDR选择的准确性。

4, 剂量递增阶段观察疗效,有疗效的剂量组进入剂量扩增阶段,以随机形式选出最终的RD。

5, 提供更为灵活的模板,在每个剂量组完成后评估,患者可以接受下一个DL治疗,或者进行随机。

上述5种方法的一些共性特征:
1. 以MTD为主要衡量标准的方法学将被淘汰,取而代之的是综合多项指标(安全性、疗效、PD、PK),在剂量递增阶段选出2个及以上的备选剂量,再通过随机选择最终的给药剂量。
2. 方案设计要有充分的弹性、高效的团队配合,以确保患者入组、治疗、数据采集、整理、分析和决策,有能力在短时间内完成。
3. 目前的剂量爬坡方法学包括rule或model-based,其原则包括:在有限的样本量下尽可能准确找到MTD、控制在MTD以下(无效)和以上(毒性)患者的数量。因此,这些以MTD为核心的方法学也需要进行相应调整,以适应新的剂量探索的要求。但无论如何,传统3+3或rolling six是比较稳健的。
回顾监管“新”要求,对RP2D的要求以及支持AA的证据级别,对以抗肿瘤药物开发为主的企业可能会产生较大影响。对RP2D的要求势必增加研究设计和执行的难度,以及需要更长的研究时间和配套资源,但长期来说一定是有积极作用的。
更重要的是关于加速批准的要求,如果三期确证性研究中的早期数据需要作为AA的支持证据,对于企业的资源要求则更高。特别是资源(人才、资金、经验、系统等)缺乏的创业型生物技术公司,恐怕要未雨绸缪,早做准备。
第三,FDA近期提到关于伴随诊断(CDx)的开发。
通常来讲,一药一伴随。
而CDx的开发有三种模式:1. 与抗肿瘤药物同步开发。其优势是通过个别研究,同时证明检测技术的可靠性(支持CDx的注册),以及该技术筛选出的患者对新治疗的获益(支持研究的开展和新药的获批)。2. 采用临床试验分析方法进行病例的生物标记物分析,后续再进行CDx的开发,例如早期研究中。3. 选择已经上市的CDx产品作为该药物的伴随诊断试剂。这种情况需要桥接研究,以证明该产品确实可以准确检出在临床研究中获益的患者群体。
无论哪种类型,基本都是一药一伴随。以KRAS为例,FDA获批的CDx产品检测包括FoundationOne CDx(Foundation Medicine)、Cobas Kras Mutation Test(Roche Molecular Systems)、Therascreen KRAS RGQ PCR kit(Qiagen)。显然,这会导致检测结果无法在不同产品之间认可,也增加了或许可以降低的开发成本。
因此,2022年底,Richard Pazdur在Friends of Cancer Research年会上表示,美国FDA正在启动一项试点项目,推出检测“最低性能标准”(minimal performance criteria)的概念。
这一概念意味着,医生们可以使用任何满足这一标准的检测,而不需要使用与药物绑定的特定伴随检测。这一概念的落地势在必行也带来更多的便利性,但同时也存在诸多困难。例如,如何保证minimal performance criteria在全球不同研究中心得以统一,谁来监管,特别是抗体相关的检测方法?
第四,FDA提出的第四个问题是关于动物研究。2022年9月29日,美国参议院通过法案,目前是取消对新药和仿制药进行动物实验的强制要求,旨在未来几年里大幅减少动物实验的使用。这标志着80多年来一以贯之的监管要求将发生了重大调整。
一直以来,FDA要求新药在进入临床研究前,需要在一种啮齿类(小鼠)和非啮齿类动物(猴子、狗)中进行毒性测试。然而,由于超过90%的新药将最终以失败告终,动物实验预测人类研究的价值有限。与此同时,伴随非动物方法的快速发展,包括计算机建模、器官芯片的开发等,让该法案落地有了科学的保证。
当然,这并不意味着放弃动物实验,但至少在非动物研究方法上开辟了新的道路。

科学、技术与创新
突破性创新的原动力来源于科学和技术。在科学技术的推动下,已经涌现出一批突破性创新药物,例如T细胞衔接器(TCE)、双抗、KRAS抑制剂、ADC、mRNA、蛋白降解、细胞基因治疗、人工智能。
T细胞衔接器(T-cell engager, TCE)
TCE是一类以CD3作为靶点的双抗类药物,在血液肿瘤中已经取得初步成功,在研热门靶点包括CD20、BCMA和GPRC5D。

Mosunetuzumab靶向CD20*CD3,EMA和FDA分别于2022年6月和12月批准其用于难治复发滤泡淋巴瘤患者。支持批准疗效数据来自90例FL患者,ORR为80%(70-88%),60%为CR,中位随访14.9个月,DOR为22. 8个月。
CRS和神经毒性是较常见AE,发生率均为39%(其中2级CRS占15%,3级占2%,4级占0.5%),严重感染占17%,其他主要为血象的下降。
Teclistamab靶向GPRC5D*CD3,EMA和FDA分别为2022年8月和10月批准其用于难治复发骨髓瘤(RRMM)患者。支持批准疗效数据来自110例RRMM患者,ORR为61.8%(52.1-70.9%),CRS和神经毒性是较常见AE。CRS发生率为72%(3级占0.6%),神经毒性57%(3或4级2.4%),ICANS占6%,其他主要为血象的下降。
从上述数据可以看出:
1)TCE治疗部分血液肿瘤是有效的 – 原因是TCE的一端定位肿瘤细胞,另一端召集了T细胞进行攻击,发挥抗肿瘤效应;
2)CRS和神经毒性是较常见的特征性毒性– 这在CAR-T中也见过,T细胞在聚集、活化、和发挥抗肿瘤效应过程中,释放细胞因子,导致CRS和神经毒性的发生。通过逐步递增给药、增加预处理激素使用、及时给与IL-6拮抗剂等可以大大降低严重事件的发生。
理论假设和临床观察到的情况基本一致,但缺乏直接的证据。
目前仍然有很多问题没有答案,例如:这些治疗反应是怎么发生的?一个肿瘤细胞需要几个抗体结合?假设抗体结合后周围缺乏足够T细胞怎么办?如果只有抗体结合肿瘤细胞会有生物学效应么?
什么类型和状态的T细胞才是需要的?治疗后B细胞清除(CD20*CD3)会引起CD4+T细胞的衰竭么?循环中的T细胞为什么没有吸干抗体?肿瘤组织可形成间质压力阻碍大分子进入,而这些TCE是怎么进去的?免疫逃逸肿瘤微环境又如何允许TCE发挥作用?

KRASG12C抑制剂
2022年12月12日,FDA加速批准adagrasib用于接受过至少一次系统性治疗后KRAS G12C突变的NSCLC患者。Adagrasib和Sotorasib在NSCLC中分别获批,标志着KRAS中的G12C位点的成药性。

Sotorasib的确证性三期研究CodeBreaK200在2022年ESMO公布数据,1:1入组345例NSCLC患者接受sotorasib(960mg)和多西他赛治疗,中位随访17.7个月,研究的主要终点mPFS分别为5.6个月和4.3个月(HR为0.66),即sotorasib可以降低34%的PFS事件的风险,1年PFS率分别为24.8%和10.1%,ORR分别为28.1%和13.2%,中位OS分别为10.6个月和11.3个月。
CodeBreaK200结果差强人意,尽管主要终点PFS提高了1.3个月,可惜未观察到生存获益。尽管由于方案允许交叉,34%的多西他赛组患者最终接受了sotorasib的治疗,方案设计是合理的。
但G12C作为已经获批的靶点,究竟对二线肺癌患者的生存有多大帮助,仍然需要观察,FDA就此已经做出新的要求。
除二线肺癌外,KRAS抑制剂的主要竞争还是在于一线肺癌适应症的开发。KRAS突变型肺癌具有高突变率、PD-L1高表达等特点,因此方案设计时可能要考虑:联合方案的选择(PD-1或化疗)、联合治疗的给药剂量、耐受性(特别是肝毒性)、以及对照组的选择(PD-1单药还是PD-1联合化疗)。
KRAS G12C突变约占肺腺癌的10-14%,在大肠癌、胰腺癌中也可检出2-3%的患者存在G12C突变。首先是sotorasib单药治疗胰腺癌的临床I/II期数据,38例患者接受治疗,8例患者获得确认的ORR(21%),24例患者为SD,中位PFS为4个月,中位OS为6.9个月。
从数字上来看,21%的ORR高于大肠癌单药10%左右的ORR率。

例如adagrasib公布的大肠癌的早期数据。研究入组44例患者接受adagrasib单药治疗,32例接受联合西妥昔联合治疗。中位随访20.1个月和17.5个月,ORR分别为19%(单药)和46%(联合组),中位PFS分别为5.6个月和6.9个月。
3-4级治疗相关AE分别为34%和16%,没有5级AE报告。数据发表在12月份的NEJM杂志。

值得一提的几个方面:
1. 2022年12月21日,adagrasib联合EGFR治疗大肠癌被FDA授予突破性疗法(BTD);
2. 机制上解释,以往KRAS突变(EGFR-RAS-RAF-ERK通路下游)的消化道肿瘤是无法用EGFR治疗的,而同时抑制上下游从机制到临床上都得到验证;
3. 既然在CRC上得到验证,那么是否可以推广到胰腺癌等肿瘤上,使用KRAS抑制剂联合EGFR治疗?如果能攻克G12D等位点,则胰腺癌的治疗势必会出现显著进展。
Adagrasib联合西妥昔单抗治疗晚期CRC的国际多中心三期研究(Krystal-10)正在进行中,国内有多个研究中心参与,可以推荐入组。
mRNA
mRNA新冠疫苗的巨大成功唤起了人们对于使用mRNA表达治疗性蛋白质的兴趣。除新冠疫苗外,使用mRNA表达VEGF来治疗心力衰竭,以及基于CRISPR-Cas9 mRNA治疗罕见遗传病的mRNA已经开始了一系列临床研究。

与mRNA疫苗不同,mRNA疗法的开发面临更多的挑战。疫苗仅需要产生少量蛋白质,随后人体免疫系统放大免疫信号,而治疗性mRNA则需要1000倍以上的蛋白质水平才能达到治疗阈值。并且,mRNA疗法需要作用于特定的通路、细胞、组织或器官。
因此需要重视靶细胞对mRNA的吸收,这取决于mRNA表达的持续时间和水平。脂质载体递送到组织中的生物利用度、循环半衰期和递送效率等。差异请参考下图。

mRNA在治疗癌症的方向上快速发展。2022年12月,Moderna与Merck在宣布了一项临床2b期KEYNOTE-942的阳性结果。
研究评估mRNA-4157(编码34种新抗原的单一合成mRNA,新抗原是根据每位患者DNA序列突变特征演算得出)和K药联合,对比K药单药,作为肿瘤完全切除后的III/IV期黑色素瘤患者的辅助治疗。
研究入组157例患者,主要终点无复发生存,治疗组降低复发或死亡风险44%(HR 0.56,p=0.0266)。这是首次随机研究证明mRNA对于癌症的治疗效应,接下来将进入三期临床。

mRNA相比其他产品,具有耐受性好、无整合进入基因组风险、无病毒(作为载体)传染的风险、容易降解从而降低安全性风险、诱导的体液和细胞免疫将激活和维持抗肿瘤疗效、生产迅速且费用低等优点。因此,社会对mRNA作为一种全新的治疗模式用于抗肿瘤领域充满期待。
简述mRNA的作用机制:1. 成药的mRNA被APC细胞呈递,MHC I类分子激活CD8+ T细胞参与细胞免疫功能,2. 交叉活化CD4+ T细胞,活化B细胞参与体液免疫。因此,该步骤可以理解为:找到特异性癌症抗原,制成mRNA药物,激活患者自身特异性的细胞和体液免疫,发挥抗肿瘤效应。

细胞基因治疗
基因治疗三要素包括核酸、载体和靶细胞。受到对疗效持久性和长期安全性风险的担忧,监管部门对基因治疗产品的要求相对特殊。
Roctavian由BioMarin公司开发的一款治疗血友病甲的基因治疗产品,这是一款通过使用AAV5病毒载体递送表达凝血因子VIII的转基因的基因疗法
2022年8月24日,欧盟批准该基因疗法有条件上市,成为首个治疗血友病甲的基因疗法。近日,BioMarin公司近期宣布了随访超过3年的三期临床数据,在接受Roctavian治疗3年和4年的患者中,其凝血因子VIII的活性平均值分别为18.8和15.2,中位数值为8.4和7.4。两组的平均年出血事件率分别为1.0和0.8,而中位值都为0.0。
与基线值相比,在治疗的第三年后,患者的平均年出血事件率减少了80%,而凝血因子VIII的使用减少了94%。在第3年时,92%的患者仍然不需要预防性治疗。预计FDA可能会很快批准。
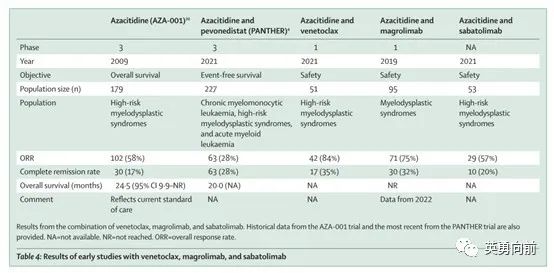
CD47和MDS
CD47是过去几年比较热门的靶点之一,特别是Forty-seven公司公布Magrolimab在AML和MDS中的良好的临床数据(CR率相比AZA单药有2-3倍的提高)并且被Gilead收购。随着这两年数据的陆续公布,对三期确证性ENHANCE研究的结果充满担忧,HR-MDS中的CR能否转换为OS?
最近一篇值得学习的文章,讨论MDS中各种新药研究的陷阱和推荐。新药的临床研究,特别是大规模确证性研究,在开始之前,深刻了解疾病的特点、分类、医学需求、终点等非常重要。
以MDS为例,这是个异质性非常大的恶性肿瘤,或者说前白血病。白血病的表现形式相对简单,但作为前白血病则表现差异性巨大。

MDS低危型的治疗目标是改善造血、延缓疾病进展,而对于高危型的治疗目标则更接近常规恶性肿瘤。然而,以往区分AML和MDS的原始细胞比例为20%(现已稍微调整),因此MDS通常的原始细胞比例都不高,CR代表检测水平下肿瘤的消失但在MDS中的意义是不同的(MDS主要表现在细胞遗传学水平的异常– 前白血病)。二期数据中的CR能否转化为OS获益呢?
文章对MDS的临床研究提出一些建议:
1)小型研究一般难以成功– 统计的力量;
2)根据以往经验,CR不应该作为批准新药的主要终点;
3)研究应该是盲态的,因此被分配到对照组可能会导致终止治疗,影响研究结果;
4)重新考虑对MDS的分类– 应纳入可靠的遗传学信息例如TP53等,至少作为分层因素加以处理。
希望此类文章更多些。
癌症数据报告以及行业进展
Cancer Statistics, 2023公布了美国癌症统计报告。2023年,预计美国发生195万癌症新发患者、60万死亡。据统计,美国的癌症死亡率从1991年来总体下降了33%,呈逐年下降趋势。

诸多因素包括生活方式的变化(减少吸烟)、癌症的早期发现和治疗、疫苗等普及。所有癌症的5年生存率从70年代的49%到近期的68%,其中存活率最高的癌症包括甲状腺癌(98%)、前列腺癌(97%)、睾丸癌(95%)、黑色素瘤(94%),而存活率最低的癌种包括胰腺癌(12%)、肝癌和食道癌(21%)。肺癌领域的治疗进展最为迅速。

2022年FDA批准37个新药,21个为小分子产品,1个siRNA,其他15个新药为蛋白类产品(单抗6个,双抗4个,ADC为1个,融合蛋白1个)。
从创新上来看,20款产品为first-in-class疗法,占比54.1%(20/37),为近十年来比例最高。审批程序上,12款获得Fast Track,13款为Breakthrough Designation,12款获Priority Review,6款Accelerated Approval。97%的产品(36/37)在PDUFA内批准,68%的产品为美国首批。
2022年批准10个抗肿瘤新药(27%),例如,1)adagrasib,治疗KRAS G12C突变的NSCLC;2)mosuntuzumab,治疗难治复发滤泡淋巴瘤;3)olutasidenib,治疗IDH1突变的难治复发AML患者;4)futibatinib,治疗FGFR2基因融合的肝内胆管癌;5)teclistamab,治疗RRMM患者。

商业预测,Nature Review Drug Discovery杂志预测2023年销量最高的10个产品,其中Pembrolizumab排名第一,预计销量238亿美元,Nivolumab排名第6销量为110亿美元。
回顾过去10年里(2011-2020年)获批的168个新药,36个(21%)的新药年销量超过10亿美元(blockbuster),其他高销量(5-10亿美元)、中销量(1-5亿美元)和低销量(1亿以下)分别为31个(18%)、51(30%)和50个(30%)。
按照疾病领域区分,肿瘤/血液占比最高达37%、其次为感染(13%)、代谢、神经系统和心血管等。拥有最多Blockbuster药企是Gilead(6),其次为BMS和罗氏均为5个产品,而Merck凭借单产品获得超额销量。


版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。





































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)