
2022-11-16 来源 : 青椒医学 ,作者青椒哥
组蛋白(histone)是真核生物体细胞染色质中的一种碱性蛋白质,和DNA共同组成核小体结构。它们是染色质的主要蛋白质组分,作为DNA缠绕的线轴,在基因调控中发挥重要作用。组蛋白修饰(histone modification)是指组蛋白在相关酶作用下发生甲基化、乙酰化、磷酸化等修饰的过程。组蛋白修饰在细胞中同DNA修饰以及组蛋白变体等表观遗传变化常被用作动态调节染色质的结构和功能。这是因为组蛋白修饰改变了底物氨基酸残基的性质,这种改变通常比突变更为重要,因为它们可能影响组蛋白结构,从而影响其功能,并且翻译后修饰在组蛋白中含量丰富,特别是在其N端尾部,它们能够在调节染色动力学和多种DNA模板化生物过程中发挥重要作用,而组蛋白修饰这些过程的失调与多种疾病的发展进程密切相关。
近年来,以组蛋白修饰为研究对象的高分文章不断涌现,而且近几年国家自然科学基金的资助项目中,组蛋白修饰相关项目数量激增。说明组蛋白修饰已成为当前生命科学/基础医学研究的一大热点。
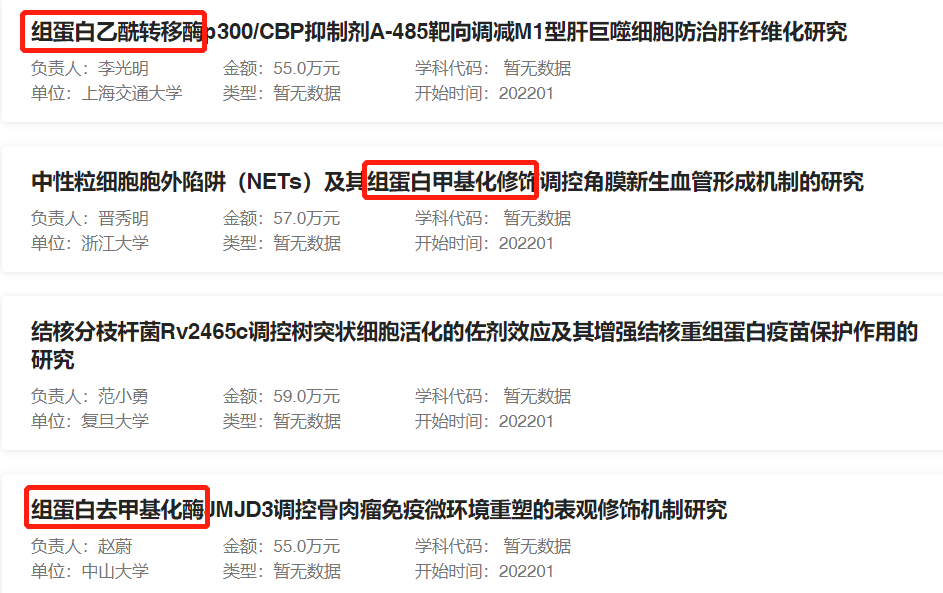
2021年组蛋白相关资助的部分项目
小编为大家整理了10篇近期发表的组蛋白修饰相关高分文章,另附每篇文章的分子机制图和原文链接,有助于了解最新的代谢重编程研究思路,感兴趣的文章可跳转具体链接研读全文。
1. Histone acetylation of bile acid transporter genes plays a critical role in cirrhosis
J Hepatol(IF 25.083)
Pub Date: 2022-04-01
分子机制:MCRS1是一个组蛋白乙酰化调节因子,通过一个先前未被描述过的SANT结构域锚定HDAC1和HISH3,在基因表达和肝脏健康的适当调节中发挥关键作用。肝细胞MCRS1的缺失通过增加胆汁酸(BA)转运蛋白基因的组蛋白赖氨酸乙酰化,在肝窦内积聚BAs,激活肝星状细胞(HSCs)上的FXR,从而诱导肝硬化。这些研究结果表明,肝成纤维细胞中胆汁酸/FXR 轴的激活是肝硬化发展的关键,该轴代表肝硬化中的中心和普遍信号事件,通过抑制组蛋白乙酰转移酶来靶向肝硬化患者的组蛋白乙酰化可能是治疗肝硬化可行选择。
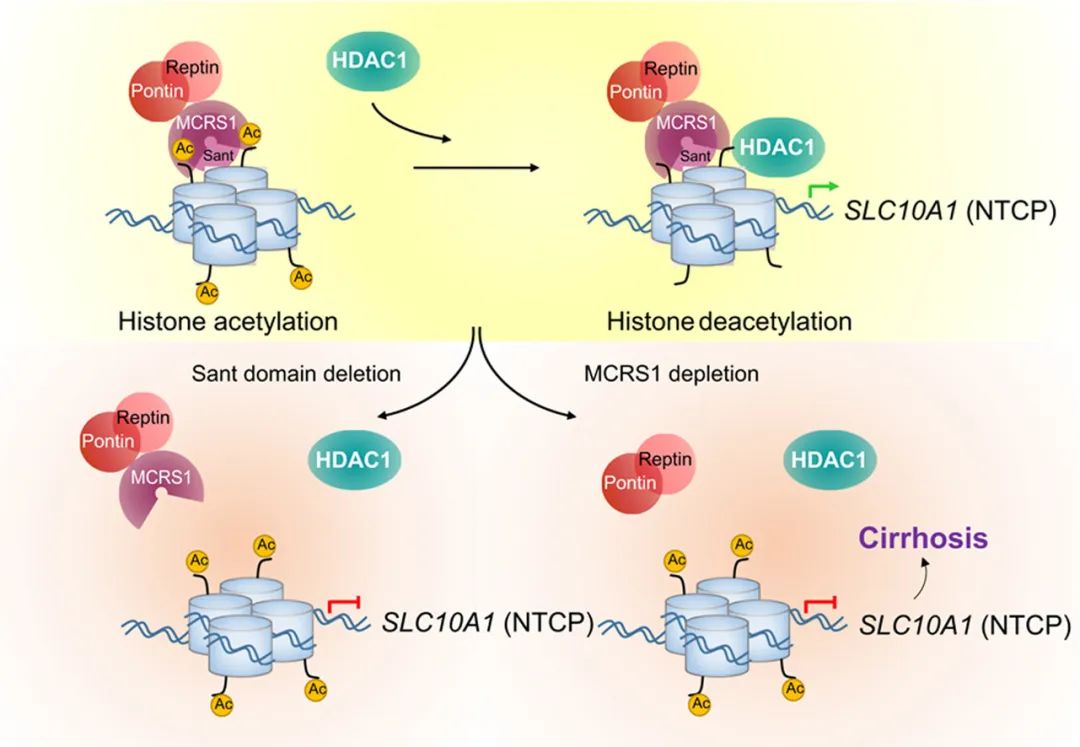
原文链接:https://www.journal-of-hepatology.eu/article/S0168-8278(21)02294-7/fulltext
2. Glucose starvation induces a switch in the histone acetylome for activation of gluconeogenic and fat metabolism genes
Mol Cell(IF 17.97)
Pub Date: 2022-01-06
分子机制:饥饿诱导(葡萄糖剥夺)会降低细胞内的乙酰辅酶A水平,并在去乙酰化酶和乙酰转移酶调控下导致乙酰化模式发生基因组水平的重构,促生长基因的乙酰化和转录受到抑制,而乙酰基团富集于糖异生与脂类代谢基因的调控区域促进其乙酰化与转录表达。这种重新分配由组蛋白去乙酰化酶 Rpd3p 和乙酰转移酶 Gcn5p(SAGA 转录共激活因子的一个组成部分)介导。这些研究结果揭示了营养胁迫下细胞通过组蛋白乙酰化的重新整合而调节转录与代谢网络的新机制及组蛋白乙酰化在饥饿条件下激活新陈代谢和生存所需的关键基因的功能。
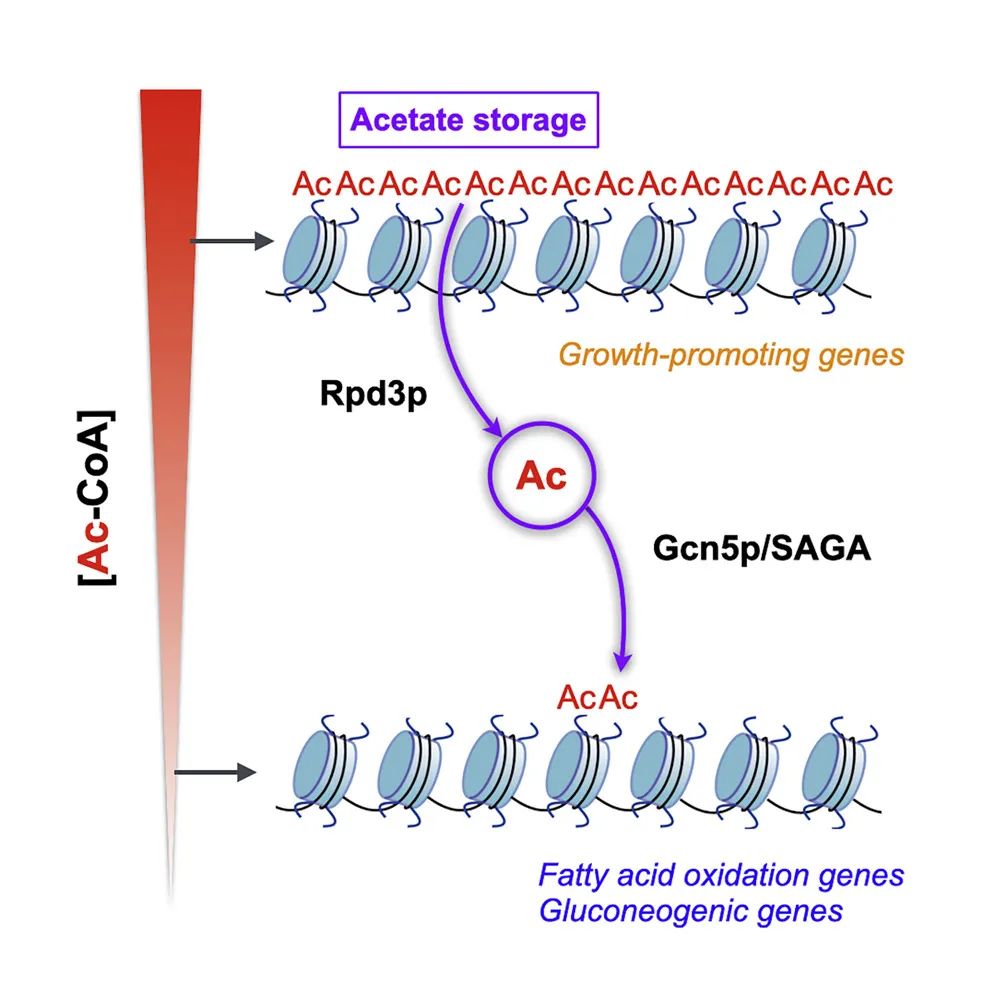
原文链接:https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)01077-7?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1097276521010777%3Fshowall%3Dtrue#relatedArticles
3. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease
Cell Metab(IF 27.287)
Pub Date: 2022-03-11
分子机制:小胶质细胞的促炎激活是阿尔茨海默病 (AD) 的标志,这一过程涉及从氧化磷酸化 (OXPHOS) 向糖酵解的转变。AD 小鼠模型 (5XFAD)小鼠和 AD 个体的脑样本中组蛋白乳酸化水平均升高,且与 Aβ 斑块相邻的小胶质细胞中 H4K12la 水平升高。这种乳酸依赖性组蛋白修饰在糖酵解基因的启动子处富集并激活转录,从而增加糖酵解活性。最终,糖酵解/H4K12la/PKM2 正反馈回路加剧了 AD 中的小胶质细胞功能障碍。PKM2 的药理学抑制减弱了小胶质细胞的活化,而 PKM2 在小胶质细胞中的特异性消融改善了 AD 小鼠的空间学习和记忆。这些研究结果表明,糖酵解/H4K12la/PKM2正反馈回路的破坏可能是针对AD 的潜在治疗方法。

原文链接:https://www-sciencedirect-com.proxy.library.carleton.ca/science/article/abs/pii/S1550413122000857?via%3Dihub
4. Loss of histone methyltransferase SETD1B in oogenesis results in the redistribution of genomic histone 3 lysine 4 trimethylation
Nucleic Acids Res(IF 16.971)
Pub Date: 2022-02-28
分子机制:组蛋白 3 赖氨酸 4 三甲基化 (H3K4me3) 是在基因启动子和 CpG 岛中发现的表观遗传标记,对哺乳动物发育至关重要。H3K4me3 甲基转移酶 SETD1B 和 MLL2 (KMT2B) 对卵子发生至关重要。H3K4me3 在 Setd1b条件性敲除 (cKO) 卵母细胞中重新分布,表现为与下调基因表达相关的活性基因启动子的丢失。并且许多区域也获得了 H3K4me3,特别是那些 DNA 低甲基化、转录无活性和富含 CpG 的区域,这是 MLL2 靶标的标志。因此,SETD1B 的缺失破坏了 MLL2 和从头 DNA 甲基转移酶之间的平衡,导致卵子发生过程中的表观遗传现象。这些研究结果揭示了 H3K4me3 在卵子发生中基因组靶向的两种不同的互补机制,SETD1B 与基因表达相关,MLL2 与 CpG 含量相关。
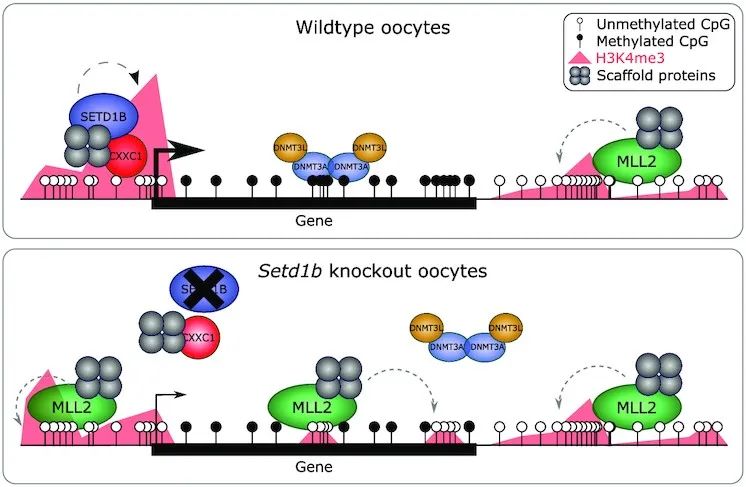
原文链接:https://academic.oup.com/nar/article/50/4/1993/6523799?login=false#334429819
5. SUMOylation of linker histone H1 drives chromatin condensation and restriction of embryonic cell fate identity
Mol Cell(IF 17.97)
Pub Date: 2022-01-06
分子机制:接头组蛋白 H1 受到 SUMO2/3 的翻译后调节,这有助于其固定在胚胎干细胞 (ESC) 中的超浓缩异染色质上。SUMO化缺失使染色质易分解,H1离开,导致全能性重新激活。此外,H1 和 SUMO2/3 共同介导全能元素的抑制。最后,我们证明抑制 H1 上的 SUMO化消除了其抑制 ESC 中全能性激活的能力。这些研究结果揭示了 H1 的 SUMO 化在促进染色质抑制和全能特性破坏中的关键作用。
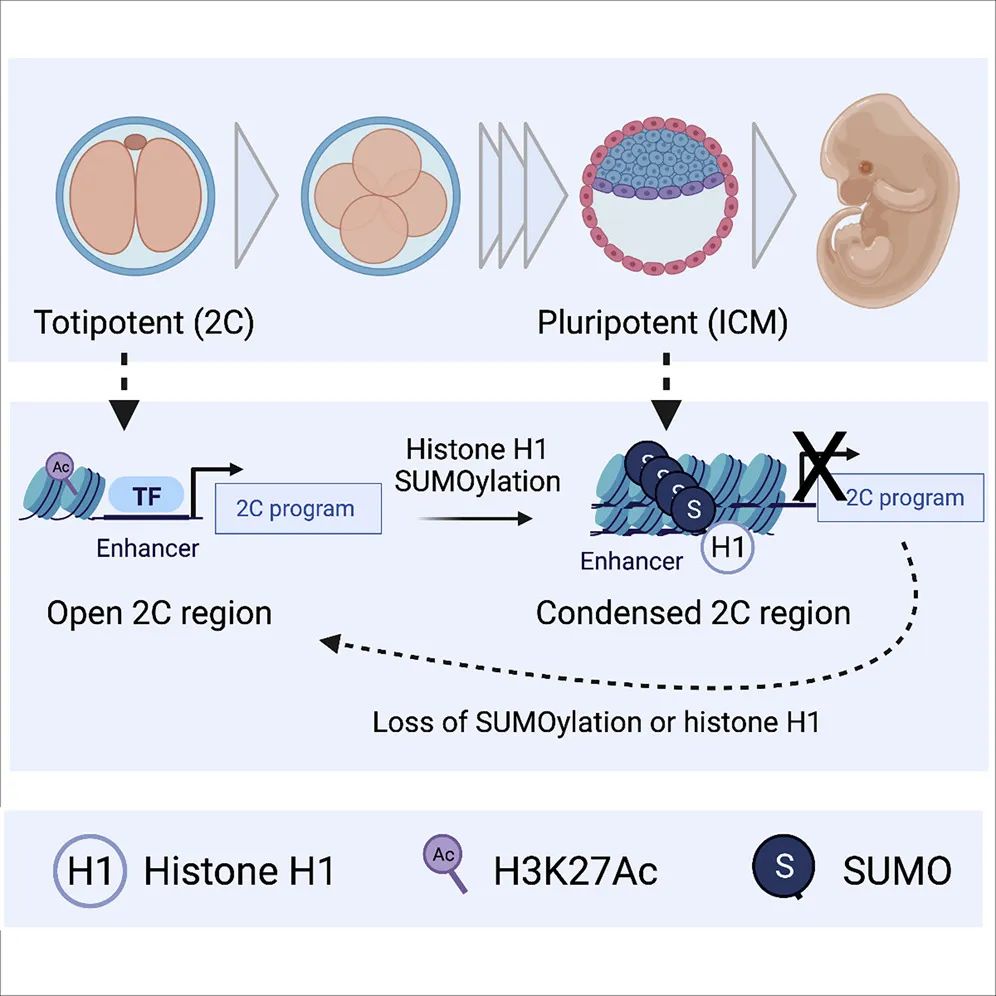
原文链接:https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)00990-4?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1097276521009904%3Fshowall%3Dtrue
2022年度国自然医学部国自然40大科研热点的中标数统计如下:
2022热点 | 2022中标数 | 2022热点 | 2022中标数 |
免疫调控 | 907 | 中性粒细胞 | 112 |
巨噬细胞 | 591 | 反馈回路 | 104 |
线粒体 | 491 | 乳酸化 | 104 |
血管功能 | 487 | 可变剪接 | 71 |
外泌体 | 470 | AI机器学习 | 67 |
自噬 | 404 | 类器官 | 67 |
铁死亡 | 337 | 炎症小体 | 62 |
干细胞 | 329 | 染色质重塑 | 58 |
代谢重编程 | 325 | 单细胞测序 | 54 |
m6A/m5C/m7G | 320 | 糖基化 | 50 |
泛素化 | 225 | 低氧缺氧 | 50 |
circRNA | 221 | 相分离 | 50 |
lncRNA | 204 | 泛凋亡PANoptosis | 42 |
细胞焦亡 | 175 | 细胞衰老 | 37 |
组蛋白 | 171 | 胞葬 | 33 |
肠道菌群 | 133 | CRISPR | 33 |
乙酰化 | 125 | 增强子 | 29 |
内质网 | 125 | 精氨酸甲基化 | 25 |
转录调控 | 112 | 迁移体 | 8 |
糖酵解 | 112 | 血管拟态 | 8 |
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。




































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)