2023-01-18

北京卫健委消息,北京市在原有苯丙酮尿症、先天性甲状腺功能减低症和先天性肾上腺皮质增生症筛查基础上,扩大新生儿遗传代谢性疾病筛查病种,将遗传代谢性疾病筛查病种由3种扩增至12种。
新增筛查病种为:枫糖尿症、甲基丙二酸血症、丙酸血症、异戊酸血症、戊二酸血症I型、3-甲基巴豆酰辅酶A羧化酶缺乏症、原发性肉碱缺乏症、中链酰基辅酶A脱氢酶缺乏症、极长链酰基辅酶A脱氢酶缺乏症。
6月1日及以后本市出生的本市常住人口新生儿,在家长知情同意情况下,可享受上述12种遗传代谢性疾病的免费筛查。
遗传代谢病是一大类高致残率和高致死率的遗传性疾病,具有种类多、病因复杂、临床异质性高、诊断困难等特点,是导致我国儿童死亡的主要原因之一,也是导致出生缺陷的重要原因。增加新生儿遗传代谢病筛查病种,将惠及更多的患儿及家庭。
苯丙酮尿症、枫糖尿症、甲基丙二酸血症三种遗传病前面已有介绍,小伙伴可翻看之前的文章:“不食人间烟火的天使--苯丙酮尿症”、“带甜味的宝宝 —— 枫糖尿症”、“甲基丙二酸尿症-儿童最常见的有机酸代谢性疾病”。
今天再随小编一起来了解一下发病率位居第二的有机酸遗传代谢病——丙酸血症。
>>>什么是丙酸血症?
丙酸血症(propionic acidemia, PA)又称丙酸尿症,是一种罕见的常染色体隐性遗传代谢病,由于丙酰辅酶A羧化酶(propionyl-CoA carboxylase, PCC)缺陷导致支链氨基酸和奇数链脂肪酸代谢障碍,使得丙酸和旁路代谢产物在体内蓄积,引起一系列生化代谢紊乱及全身多系统损害,以脑损害为主。
丙酸血症的总体发病率为1:100,000~1:50,000,不同国家和地区存在差异,我国为0.6/100,000~0.7/100,000。在我国,丙酸血症的发病率仅次于甲基丙二酸血症,是一种危害性较大的遗传代谢病。
>>>哪些基因突变可导致丙酸血症?
丙酸血症由编码线粒体丙酰辅酶A羧化酶基因PCCA或PCCB缺陷所致。丙酰辅酶A羧化酶定位于线粒体基质,由6个α-亚基和6个β-亚基组成,分别由PCCA和PCCB基因编码。α-亚基连接在β-亚基中心核外侧,含有生物素结合结构域和生物素羧化酶结构域,负责ATP水解后羧基生物素的形成;β-亚基六聚体组成PCC酶的中心核,具有丙酰辅酶A结合位点和羧基转移酶结构域,负责将羧基转移至丙酰辅酶A。
丙酸的代谢途径(PMID: 22593918)
在支链氨基酸、苏氨酸、甲硫氨酸、奇数链脂肪酸和胆固醇侧链的代谢过程中,丙酰辅酶A羧化酶在生物素的协助下,将丙酰辅酶A羧基化,生成甲基丙二酰辅酶A,后者进一步转化为琥珀酰辅酶A,进入三羧酸循环进行能量代谢。当PCCA或PCCB任一基因存在双等位基因突变时,丙酰辅酶A羧化酶活性降低或缺失,导致丙酸和丙酰辅酶A异常堆积,进而引起丙酰肉碱(propionyl-carnitine, C3)、甲基枸橼酸、3-羟基丙酸等多种中间产物在体内蓄积。
>>>丙酸血症有哪些临床表现?
丙酸血症临床表现多样,主要表现为高血氨、脑损伤和心肌病等。可在新生儿至成年任一阶段发病,无性别差异。按照发病年龄分为早发型和晚发型,早发型是最常见的类型。
早发型:1岁以内
1岁内发病,常在出生后数天至数月发病。常见症状是喂食困难、呕吐、脱水、低体温、嗜睡、发育落后、肌张力低下、癫痫、呼吸困难,若未及时治疗,病情进行性加重,出现酮症、高氨血症、代谢性酸中毒、昏迷,病死率极高,存活者多遗留智力运动障碍、癫痫等后遗症。
晚发型:1岁至成人
于1岁至成年发病,患者个体差异明显,主要表现为厌食、发育迟缓、惊厥、肌张力低下、精神行为异常等。常因发热、饥饿、高蛋白饮食、感染、药物、预防接种等因素诱发急性代谢紊乱。
>>>如何诊断丙酸血症?
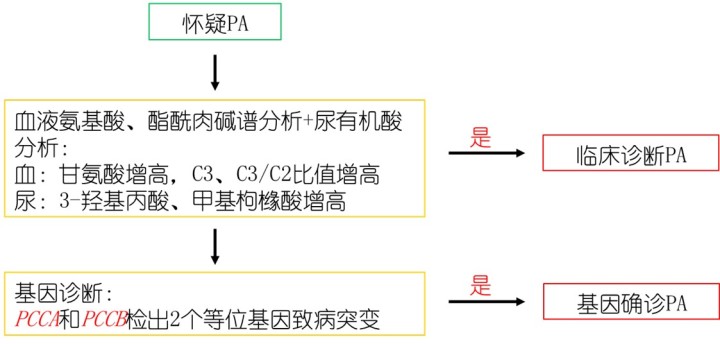
丙酸血症的诊断主要依靠液相色谱串联质谱技术测定血浆氨基酸和酰基肉碱谱水平,并结合气相色谱-质谱技术检测尿有机酸谱,最终根据基因检测确诊。
丙酸血症诊疗流程(罕见病诊疗指南2019年版)
1. 临床诊断
新生儿生后数小时到1周内出现拒乳、呕吐、嗜睡、肌张力低下、惊厥、呼吸困难、高血氨、酮症、低血糖、酸中毒等异常;婴幼儿不明原因反复呕吐、惊厥、意识障碍,严重的酸中毒、高血氨,伴有特殊的影像学异常及血液系统损害者,特别是有类似/不明原因死亡家族史时,应考虑丙酸血症。进一步行血氨基酸和酯酰肉碱谱分析提示甘氨酸水平增高,C3/C2增高;尿有机酸分析见3-羟基丙酸和甲基枸橼酸增高,可临床诊断丙酸血症。
2. 基因诊断
采用Sanger测序、靶向捕获二代测序和全外显子组测序等技术检测到PCCA或PCCB基因的双等位基因致病性突变,可明确基因诊断,并有助于患者家庭遗传咨询和产前诊断。
PCCA基因定位于染色体13q32.3,包含24个外显子;PCCB基因定位于染色体3q22.3,包含15个外显子。基因突变形式主要为错义突变,其次为插入、缺失以及剪切突变,也包含大片段缺失或/重复。其中,点突变在PCCA基因突变中约占80%,在PCCB基因突变中约占97%;大片段缺失/重复在PCCA基因突变中约占20%,在PCCB基因突变中约占3%。若测序未发现或仅发现1个等位基因突变时,应进一步进行拷贝数变异分析。
>>>如何治疗丙酸血症?
目前丙酸血症缺乏特异性的治疗方法,一经确诊应立即开始治疗,以降低致死率及致残率,避免造成持久的神经系统损害。
急性期主要以生命支持、对症治疗为主,包括限制蛋白质摄入、静脉补液和纠正酸中毒、低血糖和电解质紊乱,必要时需要进行腹膜透析或血液透析。
长期治疗以限制天然蛋白饮食为主,主要是限制缬氨酸、异亮氨酸、苏氨酸和甲硫氨酸的摄入量,并辅以左卡尼汀、甲硝唑、氨甲酰谷氨酸等药物治疗。
对于经饮食和药物联合治疗效果不佳、仍反复发生严重代谢失代偿或合并心肌病的患者,可以考虑进行肝移植。
>>>如何预防丙酸血症?
丙酸血症是较为常见的有机酸代谢病,容易发生严重代谢性酸中毒、高血氨、昏迷甚至死亡。患者的预后差异很大,取决于开始治疗的时间及长期治疗的合理性,早期诊断和规范治疗可改善患者预后。丙酸血症属于常染色体隐性遗传病,患者父母再次生育再发风险为25%。因此,应对患者及其家庭成员提供必要的遗传咨询,对高风险胎儿进行产前诊断。通过新生儿筛查可及早发现丙酸血症患者,及早治疗,减少并发症,改善预后。
[避免近亲结婚]
•丙酸血症为常染色体隐性遗传,近亲结婚会增加发病风险
[携带者筛查]
•提前预知遗传风险,降低患病风险
[产前诊断]
•先证者基因明确的家庭,通过遗传咨询和产前诊断避免丙酸血症患儿的出生
[新生儿筛查]
•是早期识别诊断丙酸血症的有效方法,早发现、早诊断、早治疗,减少并发症,改善预后
>>>参考文献
1]Propionic Acidemia. 2012 May 17 [updated 2016 Oct 6]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022.
2]Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J Inherit Metab Dis. 2021 May;44(3):566-592.
3]丙酸血症的筛查、诊断与治疗 [J] . 中华实用儿科临床杂志, 2019, 34(20) : 1531-1534.
4]欧洲甲基丙二酸血症与丙酸血症诊治指南 [J] . 中华急诊医学杂志,2019,28 (5): 560-562.
5]罕见病诊疗指南(2019年版)
6]北京卫生健康委员会http://wjw.beijing.gov.cn/
文中图片均已获版权方授权
百度浏览 来源 : 星云基因

版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。












































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)