
2022-02-22 来源 : 医药魔方Med

尽管加速审批通道最初用于HIV药物,但在过去10年间,大约85%的加速批准是在肿瘤学领域。FDA冒着风险,在药物研究中看到有希望的早期信号,例如抑制肿瘤生长的能力,最终则通过带来临床益处——改善或延长患者的生存——来获得回报。
理想很丰满,现实很骨感。2021年以来,上市后验证性试验的风险和药品成本-效益的矛盾愈发凸显,加速审批受到各方越来越多的关注。
日前,Drug discovery today杂志(IF:7.851)在线发表了中国医学科学院肿瘤医院/药物临床试验研究中心(GCP)李宁教授、王书航博士团队的研究报告,系统回顾了全球加速批准罕见肿瘤新药波澜壮阔的10年历程。
医药魔方Med经授权编译,将陆续向大家介绍美国、中国以及全球多个国家使用的加速审批路径和孤儿药资格认定政策,以及过去10年来通过这些路径批准的肿瘤药物。本文为系列报道第一篇——美国篇。
目前,FDA的四大加速审批路径旨在促进和加速新药的开发和审批,以解决严重或危及生命疾病的未满足临床需求,包括优先审评(PR)、加速批准(AA)、快速通道(FT)和突破性疗法认证(BTD)。
具体而言,FDA在1992年率先建立了优先审评的加速审批通道,在1992年和1997年分别设立了加速批准和快速通道,随后在2012年推出了突破性疗法认证。目前FDA仍在开发新的路径,以加速药物审批和促进新药研发。
优先审评
优先审评(PR)可以将上市申请的审批周期缩短至6个月,相较标准审批周期(10个月)大幅提速。FDA将在收到企业提交的NDA/BLA后的60天内予以回复是否授予优先审评资格。
加速批准
加速批准(AA)针对的是那些与获得BTD资格相似的候选药物,例如用于治疗严重疾病并且能够提供相比当前疗法更显著的临床改善。最重要的是,加速批准允许采用可能预测临床益处的合理的临床替代终点作为依据批准药物上市。例如,对于一些癌症药物,无进展生存期(PFS)是可评估的更有效的替代终点,因为总生存期(OS)的终点需要更长时间来确定,从而延误患者有可能从中受益的新疗法的批准。
然而,在将PFS获益结果外推到OS时存在一定的误差空间。因此有必要进行确证性试验,如III期双盲对照试验,以进一步确证获得加速批准的药物的疗效和安全性。这些试验必须在上市后规定的时限内进行,如果不能确定药物的疗效和安全性,药物可能会撤市。
快速通道
快速通道(FT)允许企业和监管机构之间密切互动。这些互动为企业提供了缩短用于证明药物安全性和有效性所需临床试验周期的可能,并确保企业能够及时完整地准备所有必要的数据和文件,以满足监管审批的要求。对于获得快速通道资格的药物,可以分阶段滚动提交上市申请资料,而不用等到所有数据资料都完备时再提交上市申请,可以让监管机构提前动态审评上市资料。
突破性疗法认证
突破性疗法认证(BTD)授予用于治疗严重或危及生命的疾病并且有初步临床证据显示相比当前标准疗法具有治疗优势的药物。鉴于该认定仅基于初步数据,因此BTD不能保证在未来的临床试验中获得积极结果或成功获批。随着更多数据的收集,BTD药物可能不再显示出优于当前药物的优势,则会被取消该认定以节省审评资源。BTD的申请需要跟新药临床试验申请(IND)同时提交或不晚于II期会议,并且这些数据必须与当前疗法或有效的历史数据直接比较来证明候选药物的优效性。获得BTD的药物会得到监管机构更密切的研发指导和支持,包括滚动审评、深入沟通、临床试验设计指导等。
孤儿药资格认证
孤儿药资格认证在确保有潜力的新药完成审批过程中也发挥着重要作用。美国是第一个专门针对孤儿药颁布法案的国家,旨在促进治疗罕见疾病或适应症的药物的开发。美国的《孤儿药法案(1983)》鼓励制药公司开发和上市一些治疗患者人数不到20万人的疾病的药物。在美国,开发这些药物可能无法从市场中盈利。《孤儿药法案》的颁布使得孤儿药的申请大幅增加。
孤儿药资格认证并不是加速审批路径,但是许多被认定为孤儿药的药物也同时有资格获得加速审批资格。有许多国家的监管机构制定各种激励措施来促进孤儿药的开发,包括从早期研发的帮助到上市后的支持。在2011-2020年十年期间,在美国被认证为孤儿药并且获批的79种肿瘤药物中,74种(93.7%)被授予至少一项加速审批资格,超过一半(40/79,50.1%)以加速批准方式获批。
其他
同时,也有其他一些路径或资格认定旨在加快候选药物的开发或注册,如孤儿药资格或再生医学先进疗法(RMAT)认定。除这些路径外,为了满足肿瘤治疗药物的巨大需求,FDA肿瘤学卓越中心(OCE)于2018年启动了肿瘤药物实时审评(RTOR)。RTOR的目标是对上市申请做出高效审批,以确保患者尽早获得安全有效的“新疗法”。此路径下提交的上市申请必须符合特定临床试验设计和开发的标准,例如明显优于其他治疗方法的证据,清晰的研究设计和易于解释的研究终点。
首个依照RTOR路径审批的药物是2018年提交NDA的Kisqali (利柏西利)。RTOR路径可以有效缩短审评时间;例如,Piqray (阿吡利塞)是第一个通过RTOR路径获批的新分子实体(NME),比原计划的目标批准时间提前了三个月。目前,该路径仍在试点,申请人可自愿参与。
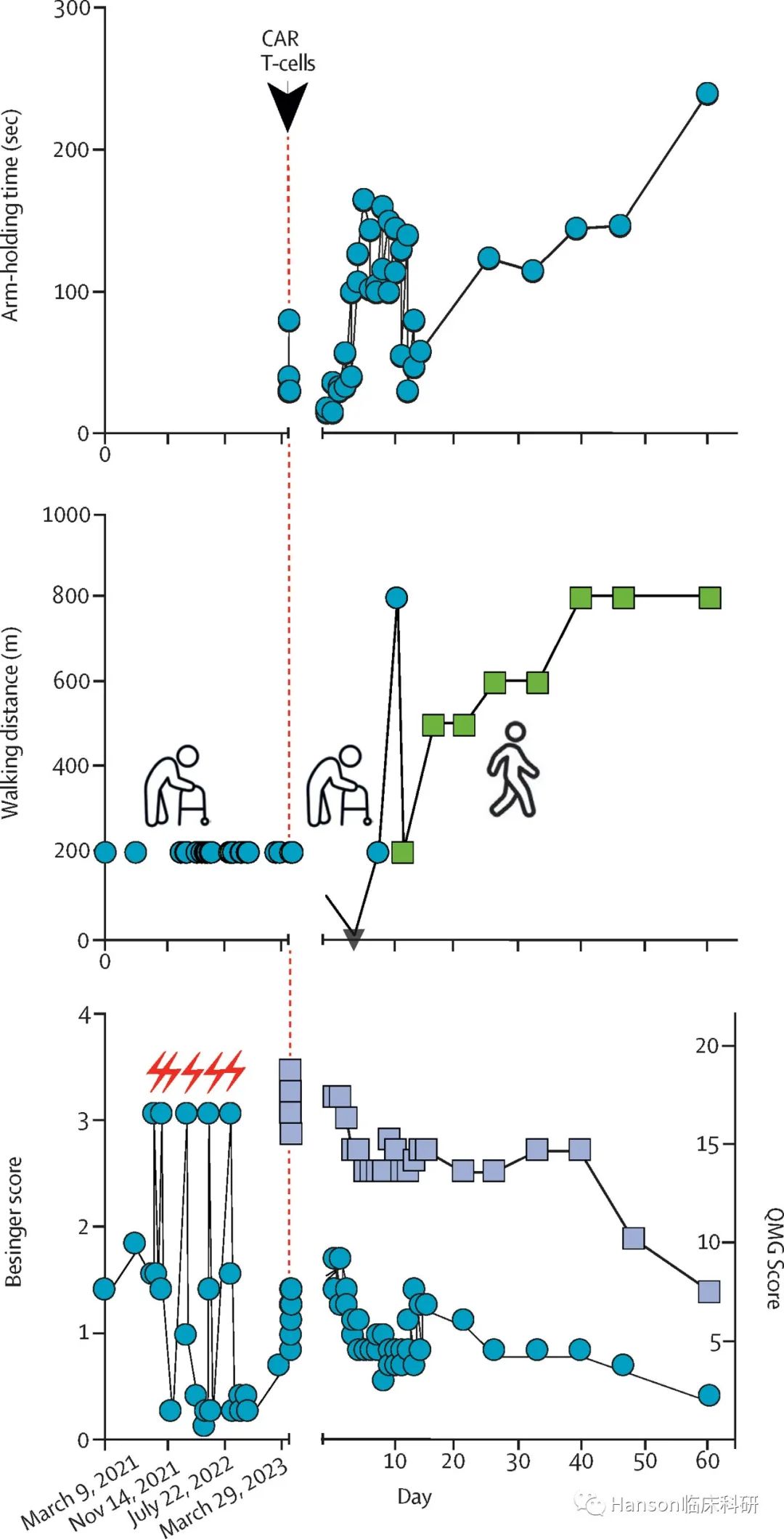
2011-2020年,美国FDA共批准410个NME,每年批准数量平均增长6.5%(图1)。超过四分之一的NME是用于癌症治疗,其中非小细胞肺癌是最常见的适应症(占13%),其次是乳腺癌、多发性骨髓瘤和黑色素瘤(分别占12%、9%和8%)(图2)。
在2011-2020年间,每年以至少一种加速审批路径获批的的药物数量占比为48.1%~72.9%。有14款药物获得了全部4种加速审批资格,这14款药物是最有希望的突破性疗法,用于治疗之前少有关注的疾病或者具有新的机制和结构。其中有12个是肿瘤药物,包括6款用于恶性血液肿瘤,6款用于恶性实体瘤,另外2款用于镰状细胞病和结核病(表1)。
加速批准允许药物基于替代终点获批
在2011-2020年间,有45.6%的药物是以加速批准的方式获批,其中约80%为肿瘤药物。在加速批准的肿瘤药物中,43款(91.5%)是基于总缓解率(ORR)数据获批上市,包括只凭借完全缓解(CR)数据,或凭借ORR加上缓解持续时间(DoR) 以及其他替代终点(包括慢性髓性白血病的主要细胞遗传学缓解和淋巴瘤的CR),这些药物仍需进行确证性临床试验。
1992-2017年,有40%的加速批准药物在加速批准上市后未如期完成上市后确证性试验,5个因为在确证性试验中缺乏疗效而被撤市,包括2011年用于治疗乳腺癌的贝伐珠单抗和2005年用于治疗乳腺癌的吉非替尼。另外有3种药物因安全性问题或缺乏确证性试验数据而被FDA撤市。还有更多的加速批准药物被企业自愿申请撤回了适应症或者正在跟FDA沟通,包括几款PD-1/L1抗体。最近,几款PD-1/PD-L1药物因确证性试验失败而被企业自愿撤回了适应症或被FDA终止审批。
自2012年推出BTD以来,FDA收到的BTD申请数量稳步增加,通过率也逐渐提高。相比而言,开发肿瘤适应症的药物更有可能被授予BTD资格(肿瘤适应症47/94,非肿瘤适应症49/257)。在获得BTD的47个肿瘤NME中,16个(34%)也同时获得了加速通道资格,34个(72%)进入加速批准通道。相比之下,40个未获得BTD的药物中,仅有9个(23%)进入加速批准通道。对于获得BTD的肿瘤药,从IND至获批的时间为24.8至257.1个月,中位数为74.8个月。
加速审批路径,尤其是BTD、AA和PR,显著提升了肿瘤药物从IND到NDA批准的开发速度。以BTD为例,获得BTD资格的药物从IND至NDA的平均时间比未获得BTD资格的药物快18个月。不同的加速审批路径中也观察到非冗余效应(图3)。
对于获得全部4种加速审批资格的肿瘤药物,从IND至NDA批准的中位周期为93.7个月(范围:32.8-122.9个月)。从获得BTD资格到NDA批准的中位周期为18.9个月(范围:0.5-51.3个月)。从NDA申请至NDA批准的中位周期为5.4个月(范围:2.4-23.5个月)。大多数药物(26/34)在NDA提交后8个月内即获得上市批准,只有4种药物历时超过10个月才获得批准。
总的来说,美国FDA在加速药物开发和审批方面做出了优秀的示范,在过去几十年中致力于推行新的加速审批路径,以满足日益增加的未满足临床需求。FDA施行时间最短的突破性疗法认定已成为行业和研究者确定最有潜力早期候选药物的风向标。
未来,随着全球药品注册和监管审查路径的不断推进,以及强调通过增强国际信任的方式以加速审批路径缩短审批时间,相信可以让更多的药物更快来到患有严重疾病(包括恶性肿瘤)的患者手中。
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。



































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)