靶向治疗
EGFR-TKI耐药
1. 一项回顾性研究:EGFR C797X突变作为奥希替尼耐药机制的真实世界现状
2. 一项研究:I期,BBT-176(四代),仍需努力
3. SAVANNAH研究:II期,EGFR-TKI耐药(MET扩增),赛沃替尼+奥希替尼ORR高达52%
EGFR 20ins
4. WU-KONG1/2/6研究:后线,I/II期,舒沃替尼(DZD9008),ORR高达52.4%
KRAS G12C
5. CodeBreaK 100/101研究:一线,I期,sotorasib+免疫,ORR为29%,毒性较大
6. 一项研究:后线,I期,Sotorasib+SHP2抑制剂,初显疗效
NTRK
7. 两项临床试验:后线,拉罗替尼,OS为40.7个月
TROP2(针对驱动基因阴性患者)
8. TROPION-Lung02研究:Ib期,TROP-ADC+K药±铂类,疗效和安全性令人鼓舞
免疫治疗
新辅助治疗
9. NADIM II研究:II期,O药+化疗 vs 化疗,pCR、PFS、OS均获益
辅助治疗
10. IMpower010研究:III期,化疗+T药 vs 化疗+BSC,OS有获益趋势
III期不可切除
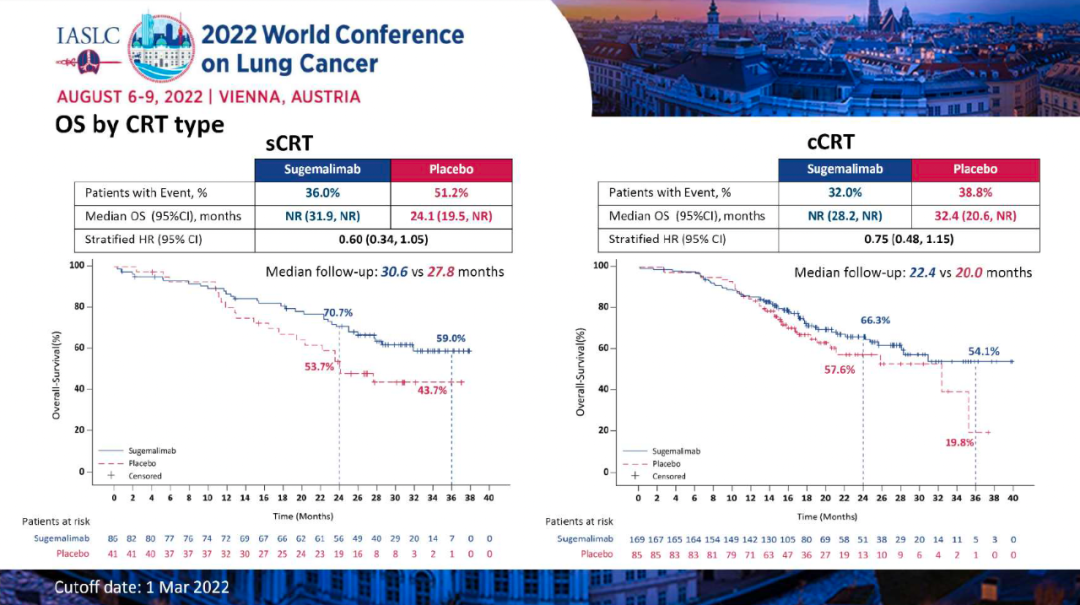
11. GEMSTONE-301研究:III期,同步/序贯CRT,舒格利单抗 vs 安慰剂维持,PFS获益
晚期免疫治疗
12. 一项汇总分析:完成35周期K药治疗,进展后接受第二疗程K药,仍具疗效
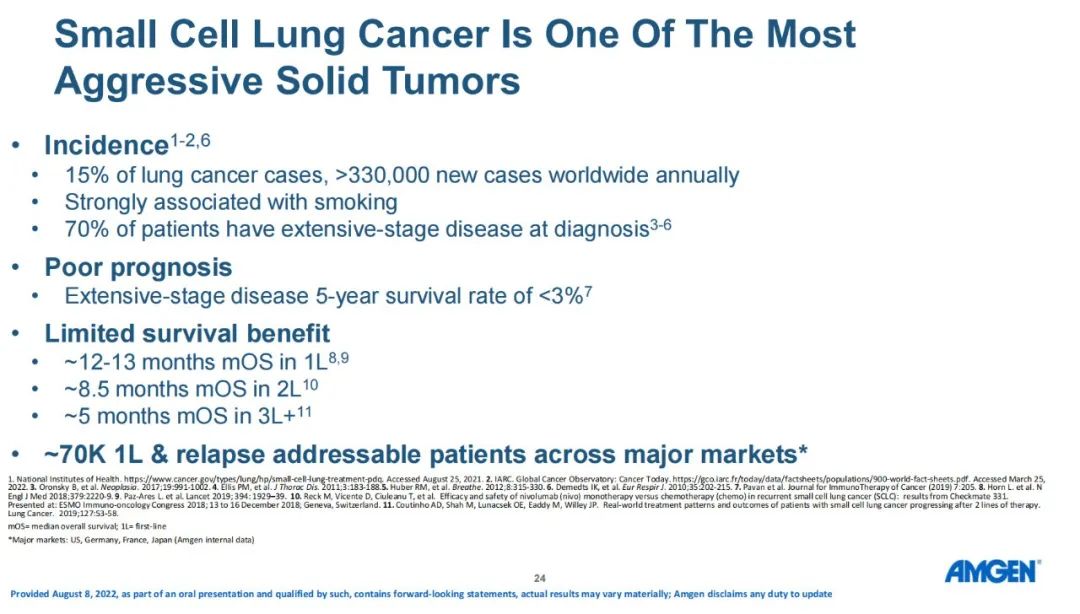
小细胞肺癌
13. DeLLphi-300研究:后线,I期,AMG 757,ORR为23%,OS为13.2个月
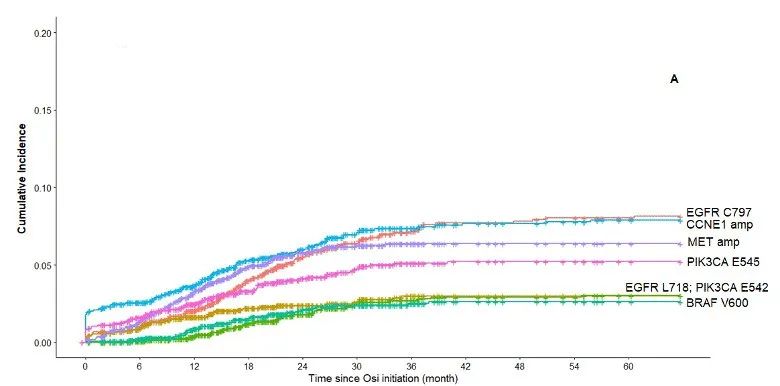
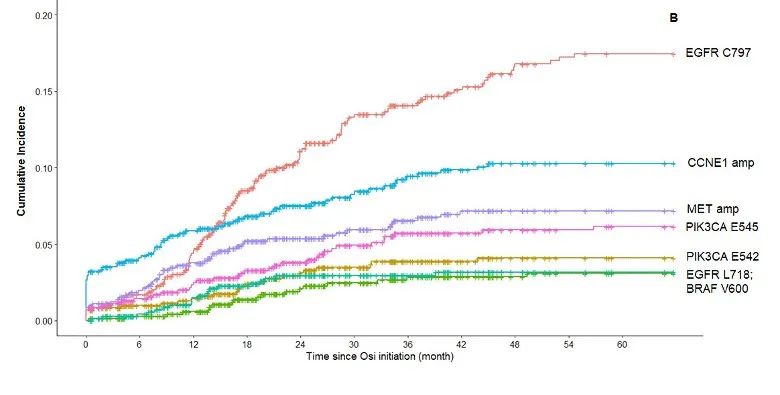
第三代EGFR-TKI 奥希替尼显著改善了EGFR突变NSCLC患者的临床结局。但奥希替尼耐药问题不可避免,EGFR C797X突变是主要机制之一。研究人员利用Guardant INFORM真实世界临床基因组数据库评估EGFR C797X和其他耐药驱动因子在EGFR突变NSCLC中的真实世界检出率和生物标志物共存情况。研究设计:这是一项回顾性观察性研究,在一个具有全国代表性的临床基因组数据库中进行,该数据库覆盖了超过174,000例晚期癌症患者。在2014年7月至2021年6月期间具有全面的ctDNA结果和相关的临床信息。通过Guardant360 (G360)液体活检纳入具有EGFR 激活突变的转移性 NSCLC 成年患者。研究结果:在71,430例NSCLC患者中,9,306例有EGFR外显子19缺失或L858R,其中1337例接受奥希替尼一线治疗,713例接受二线治疗。在接受奥希替尼一线治疗的患者中,MET和CCNE1脱靶扩增是开始奥希替尼治疗后第一年最常见的早期耐药机制,但随后变得更加罕见,而EGFR C797X靶向突变随着时间的推移显著增加,并且成为奥希替尼治疗12个月后患者中记录的最常见突变。MET、CCNE1扩增、EGFR C797X检测的中位时间分别为10.5个月、9.1个月和16.8个月。开始奥希替尼一线治疗后5年,MET、CCNE1扩增、EGFR C797X突变的累积发生率为6.4%、7.9%、8.0%,38个月后EGFR C797X突变累积发生率超过MET和CCNE1扩增。开始奥希替尼二线治疗后5年,MET、CCNE1扩增、EGFR C797X突变的累积发生率分别为7.2%、10.3%和17.5%,19个月后EGFR C797X突变累积发生率超过MET和CCNE1扩增。在一个月内停用奥希替尼的患者的子集分析中,G360检测显示C797X 的累积发病率较高。小 结:EGFR C797X是一线和二线奥希替尼治疗后最常见的获得性在靶或脱靶耐药突变,分别为8.0%和17.5%,超过了奥希替尼使用第一年后MET和CCNE1的早期脱靶扩增。研究结果表明,需继续研发下一代EGFR-TKI以应对EGFR突变NSCLC患者C797X驱动的耐药性。EGFR-TKI 是 EGFR 突变晚期 NSCLC 的标准治疗方法。但导致EGFR-TKI耐药机制众多,其中EGFR C797S突变最为常见。目前对于携带 EGFR C797S 耐药突变的患者尚未获批相关药物。BBT-176 是一种可逆的ATP竞争性抑制剂,其设计旨在抑制携带C797S突变的EGFR,并在细胞和动物功效模型中显示出低纳摩尔 IC50 值。方法:1 期研究旨在确定PK参数、安全性、推荐的 2 期剂量 (RP2D) 并探索BBT-176的疗效。研究纳入携带 EGFR 突变且既往接受过至少一种 EGFR TKI 治疗的患者入组,并通过影像学研究进行检验,每6周接受一次 Guardant 液体活检。BBT-176 从 20 mg 至 600 mg 每日一次连续口服给药,直至疾病进展或不耐受。采用贝叶斯线性回归模型指导剂量递增。允许患者内剂量递增至下一剂量水平。研究结果:截至2022年5月23日,共有25例患者接受了BBT-176治疗,其中只有8例携带C797S突变。25例患者中1例出现PR【这位PR的患者只携带19del,不过在接受BBT-176治疗的36周内已经再次进展】,确定的ORR仅为4%;5例患者仍保持疾病稳定,SD为20%【包含PR 1例】。5例患者退出研究,8例(32%)仍在接受治疗。安全性:24%的患者经历至少1次剂量降低,主要发生在接受320 mg(QD)及以上的患者中。接受320 mg(QD)及以上剂量的患者出现了3级及以上治疗相关不良事件,最常见的不良事件(≥10%)包括恶心、腹泻、呕吐、厌食、便秘、消化不良和血小板计数下降,有患者因腹泻或恶心而停止给药。
小 结:BBT-176连续每日给药的耐受性良好,毒性可管理。通过对患者进行分子选择和液体活检的动态监测,BBT-176的有效性可能进一步增强。计划在 RP2D 下进行进一步探索 (NCT04820023)。
SAVANNAH研究:是一项正在进行中的随机、单臂、全球II期临床试验(n=294),旨在评估赛沃替尼与奥希替尼联合疗法治疗既往曾接受奥希替尼治疗后疾病进展的EGFR突变、MET扩增或过表达的局部晚期或转移性非小细胞肺癌患者中的疗效。患者按300毫克或600毫克每日一次,或300毫克每日两次的剂量接受了赛沃替尼的给药治疗,联合奥希替尼80毫克每日一次的剂量治疗。主要终点是ORR。关键次要终点包括PFS、DoR和安全性。所有参加SAVANNAH研究入组筛查的患者均在奥希替尼治疗后疾病出现进展,其中62%患者的肿瘤伴有MET过表达和/或扩增,超过三分之一(34%)的患者符合定义的高MET水平阈值。该分析中的所有患者(n=193)至少为IHC50+和/或FISH5+,并在接受奥希替尼单药治疗疾病进展后,在80mg每日1次泰瑞沙治疗基础上加入300mg每日1次赛沃替尼。研究结果:1)奥希替尼+赛沃替尼治疗既往接受奥希替尼治疗后疾病进展、伴有高MET水平(定义为IHC90+和/或FISH10+)的EGFR突变的非小细胞肺癌患者的ORR为49%(95% CI,39-59%)。2)其中,在未接受过化疗的高MET水平的患者中ORR为52%(95% CI,41-63%);3)在未显示高MET水平的患者中,ORR则为9% (95% CI,4-18%)。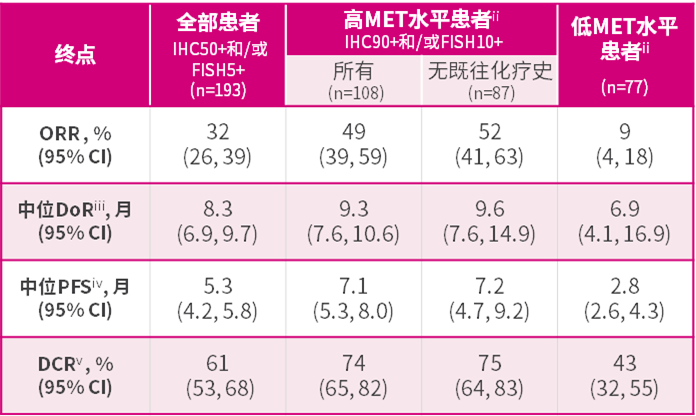
i. 分析数据截止日:2021年8月27日;ii. 亚组分析未包括八名检测结果无效或缺失的患者安全性:奥希替尼和赛沃替尼联合疗法的安全性特征与已知的联合疗法及各单药治疗的安全性保持一致。没有发现新的安全性问题。在该项分析中,少于半数(45%)的患者曾经历3级或以上不良事件,其中最常见的包括肺栓塞、呼吸困难、中性粒细胞计数减少和肺炎。13%的患者发生与赛沃替尼治疗相关的导致停药的不良事件。
患者招募丨DZD9008治疗EGFR 20号外显子插入突变的局部进展或转移性非小细胞肺癌
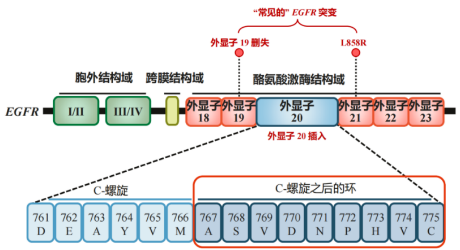
EGFR 20号外显子插入突变会改变EGFR蛋白“ɑC-螺旋”及“C-螺旋之后的环(即磷酸结合环,P-环)”的构象,形成空间位阻、导致药物结合口袋的变小,导致EGFR 20号外显子插入突变体与野生型EGFR的结合模式和亲和力更为类似,降低了传统1-3代EGFR-TKI的选择性。因此,携带EGFR 20号外显子插入突变的患者对传统1-3代EGFR-TKIs治疗不敏感。
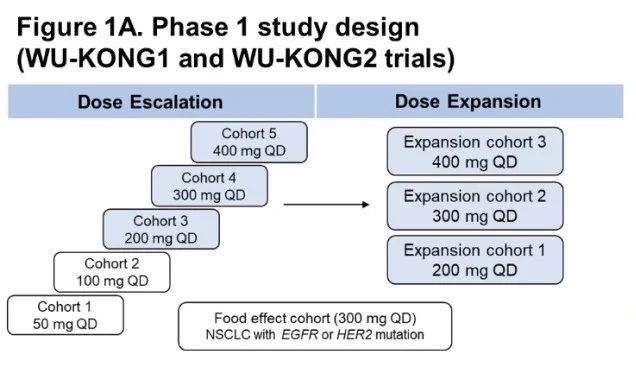
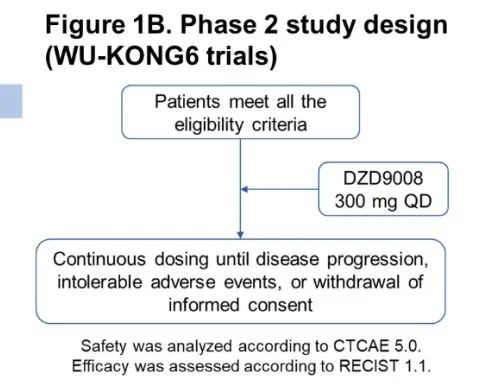
舒沃替尼(DZD9008)是一款口服、不可逆、针对多种EGFR突变亚型的高选择性EGFR-TKI,其首选适应症为治疗EGFR 20号外显子插入突变NSCLC。本次WCLC会议上报道的更新数据来自舒沃替尼三项1/2期多中心临床研究(WU-KONG1、WU-KONG2和WU-KONG6)的汇总分析。
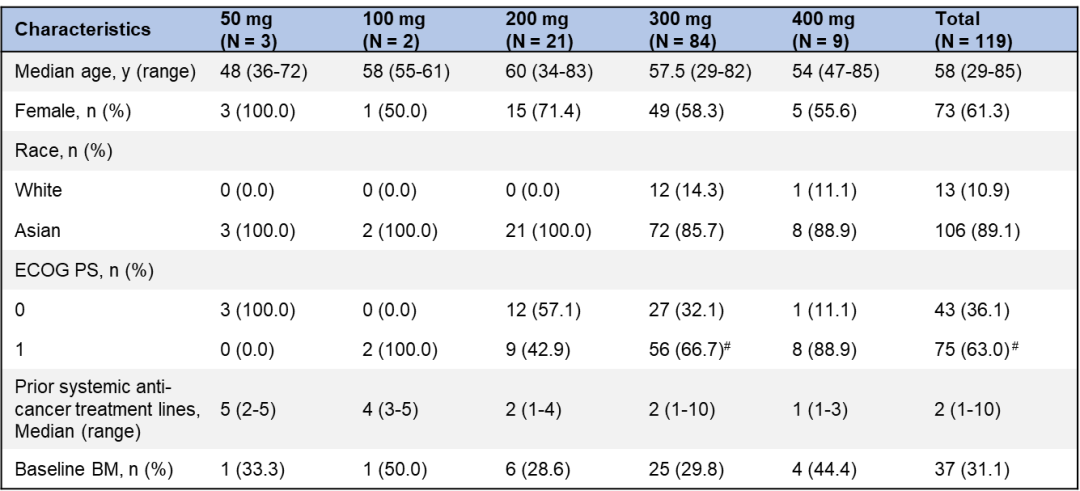
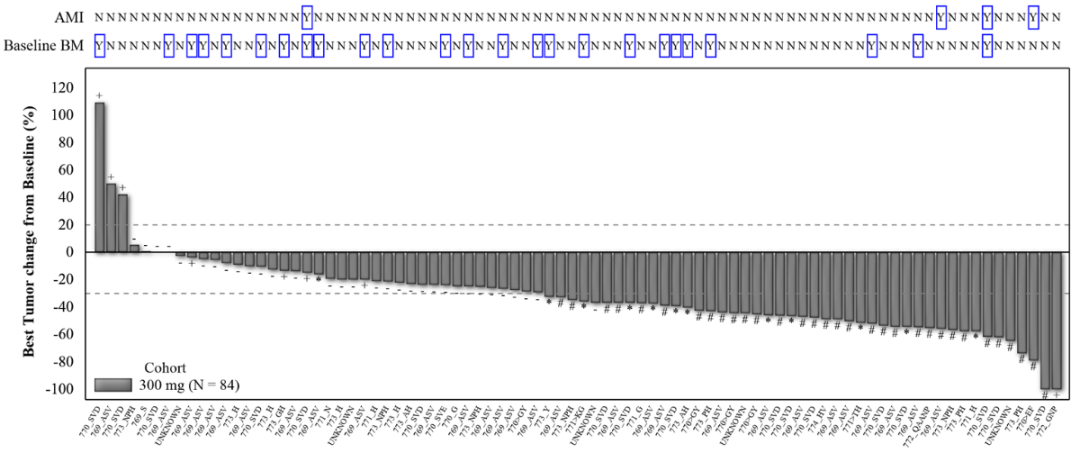
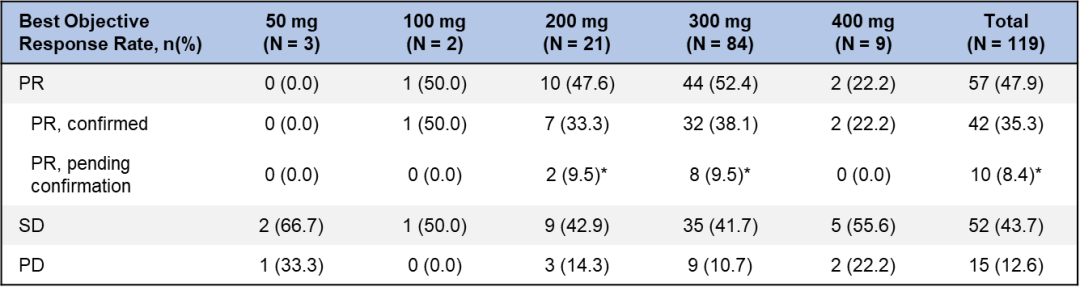
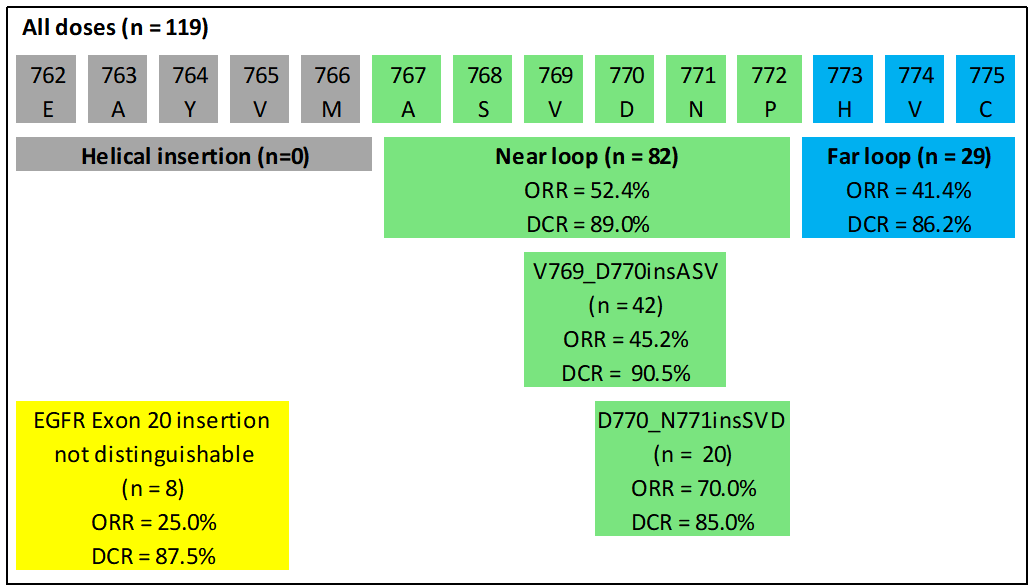
WU-KONG1研究为国际多中心研究;WU-KONG2、6为国内多中心研究研究结果:截至2022年4月30日,共有119例经化疗失败的、EGFR exon20ins突变型晚期NSCLC患者纳入疗效分析集。基线特征,BM:brain metastasis,脑转移;#1例患者(1.2%)数据散失1)84例接受舒沃替尼RP2D剂量(300 mg QD)治疗的患者,ORR高达52.4%;2)舒沃替尼对基线伴有脑转移的患者ORR达44%,表现出良好的抗肿瘤活性。左图:靶病灶大小变化的最佳百分比;AMI: Amivantamab。右图:舒沃替尼抗肿瘤活性,*: 患者仍在接受舒沃替尼治疗,疗效评估结果待确认。
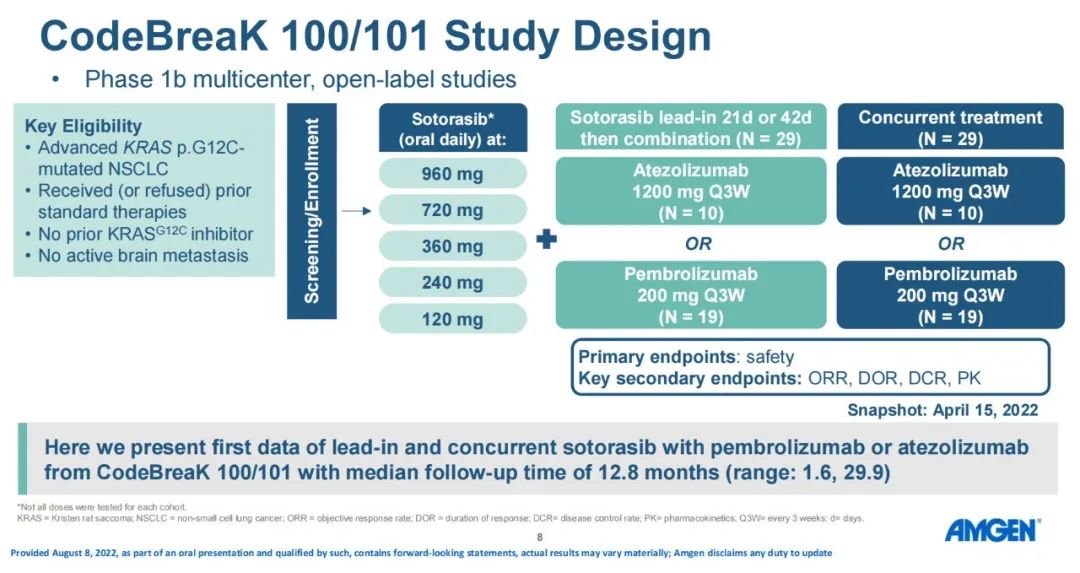
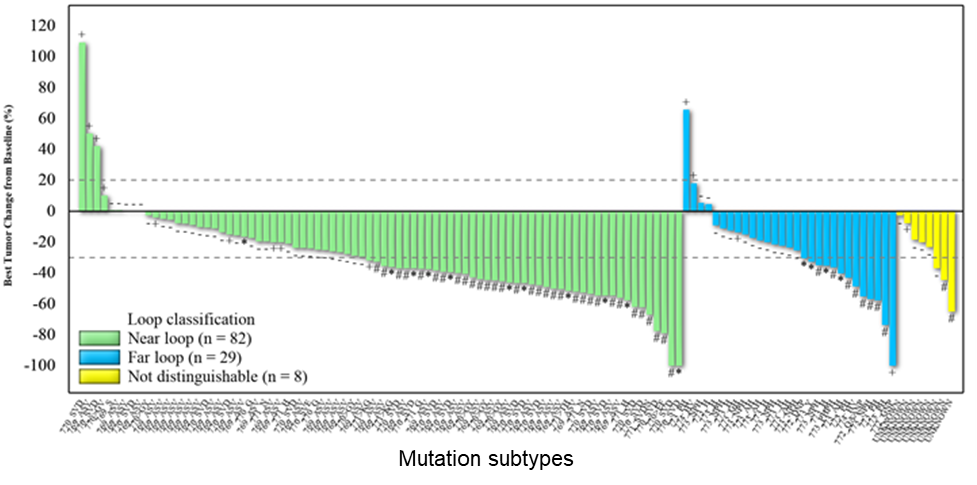
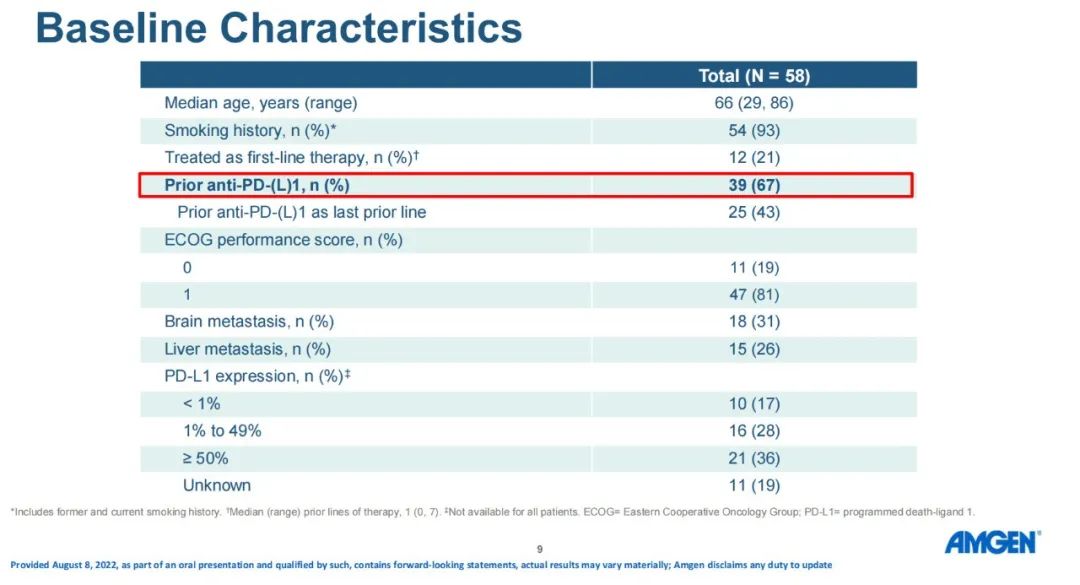
3)分析共纳入约30种EGFR exon20ins突变亚型,不论插入突变发生的位置,携带EGFR exon20ins突变的患者均能从舒沃替尼的治疗中获益【有研究提示近环端对EGFR-TKI敏感性优于远环端】。安全性:截至2022年4月30日,共238例EGFR或HER2突变的晚期NSCLC患者纳入安全性分析集。安全性数据较前进一步成熟。整体而言,舒沃替尼安全性良好,常见不良反应类型与传统EGFR-TKI类似,以腹泻、皮疹为主,且绝大多数为1-2级不良反应,临床可管理且可恢复。CodeBreaK 100/101研究:是一项Ib期多中心、开放标签的剂量探索研究,评估了“KRAS G12C抑制剂sotorasib+免疫”治疗携带KRAS G12C突变的晚期非小细胞肺癌(NSCLC)患者的疗效和安全性。研究纳入58例既往未接受过KRAS G12C抑制剂治疗的携带KRAS G12C突变的NSCLC患者,分为12个队列,接受不同剂量的Sotorasib(120-960 mg qd)联合每三周一次的静脉注射帕博利珠单抗(200 mg)或阿替利珠单抗(1200 mg)治疗,直至出现不可耐受的毒性或疾病进展。研究的主要终点是药物的安全性,关键的次要终点是ORR、DoR、DCR和药代动力学(PK)。
研究设计
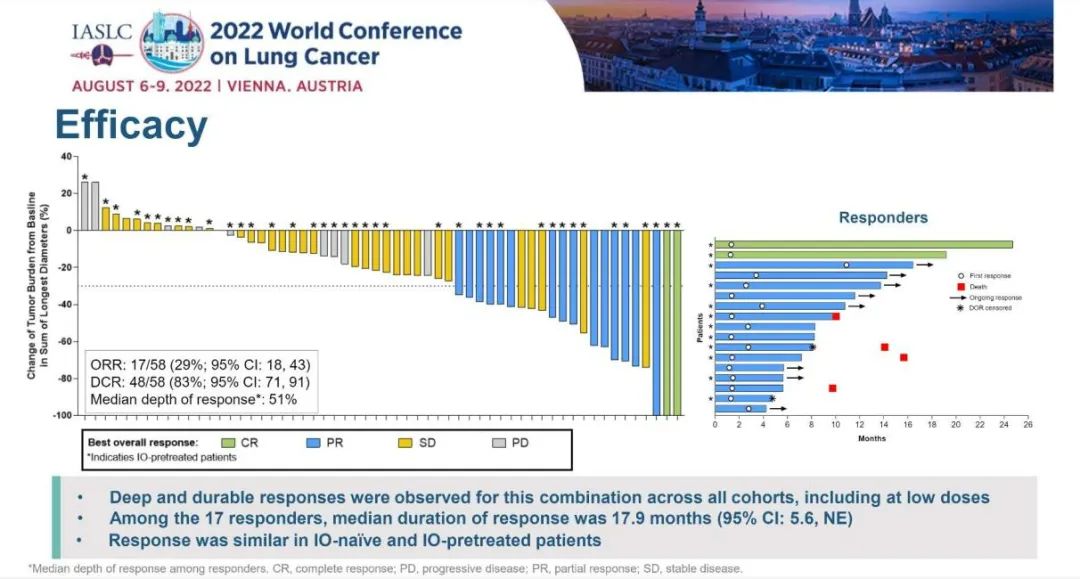
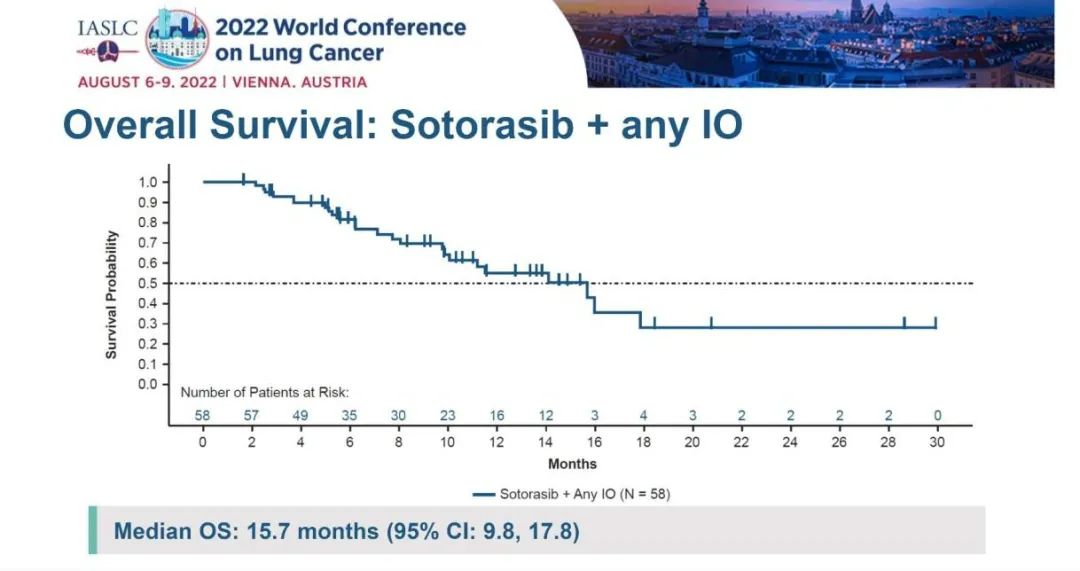
研究结果:在12.8个月的中位随访后,ORR为29%,其中的5名患者的DoR超过10个月,另8名患者仍处于持续缓解状态。另外,Sotorasib+帕博利珠单抗Lead-in队列和Concurrent队列的ORR分别为37%和32%;而Sotorasib+阿替利珠单抗ORR均为20%。DCR为83%,中位OS为15.7个月。
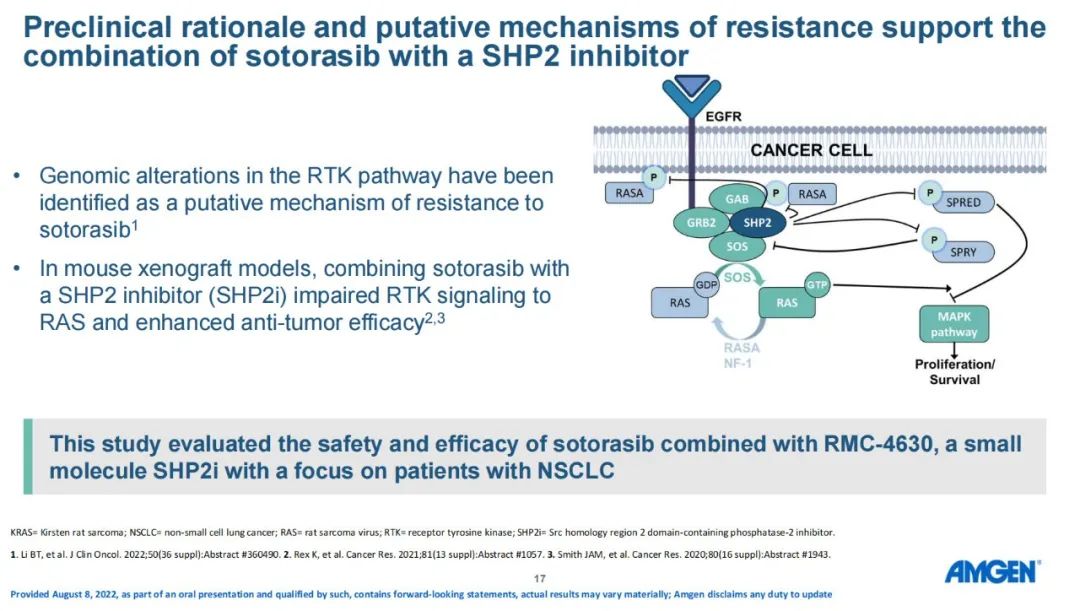
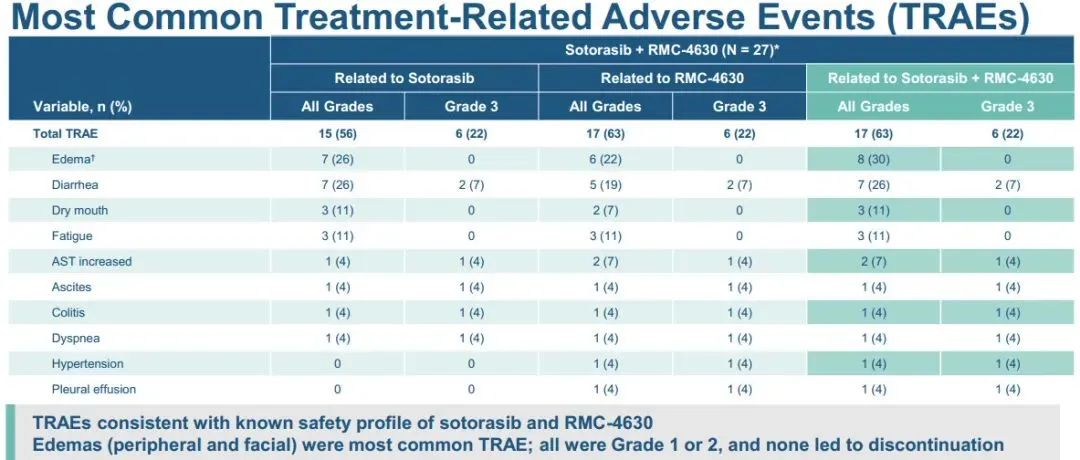
安全性:常见的3-4级治疗相关不良反应(TRAE)发生率为59%,主要是肝毒性,表现为ALT和AST水平升高。Sotorasib组合免疫疗法发生3-4级TRAE的概率高于Sotorasib单药疗法。另外,与Concurrent队列相比,Lead-in队列表现出持久的临床活性,其停药率和3-4级TRAEs发生率较低。【在试验中,同时使用sotorasib和免疫药在多名患者中导致3级以上的不良反应,主要表现为肝脏毒性。这让研究人员决定更改给药方式,先对患者进行sotorasib治疗21天或42天,然后添加PD-1或PD-L1抑制剂。试验结果显示,使用新的给药方式(lead-in)可以降低肝脏毒性不良反应。97%的肝脏毒性事件在接受皮质类固醇治疗、给药方案调整或中止治疗后获得消除。】Sotorasib是一种特异且不可逆的KRAS G12C抑制剂,单药治疗KRAS p.G12C突变实体瘤已显示出临床活性。在CodeBreaK100 PH1/2试验中,受体酪氨酸激酶(RTK)的基因组改变被确定为sotorasib常见假定耐药机制,强调了sotorasib与上游RTK信号抑制剂联合的潜在作用。在小鼠异种移植模型中,sotorasib和SHP2抑制剂(SHP2i)联合应用会削弱RAS的RTK信号传导,并增强抗肿瘤疗效。本次会议研究者报道了“sotorasib+小分子SHP2i RMC-4630”的首个安全性和有效性数据。
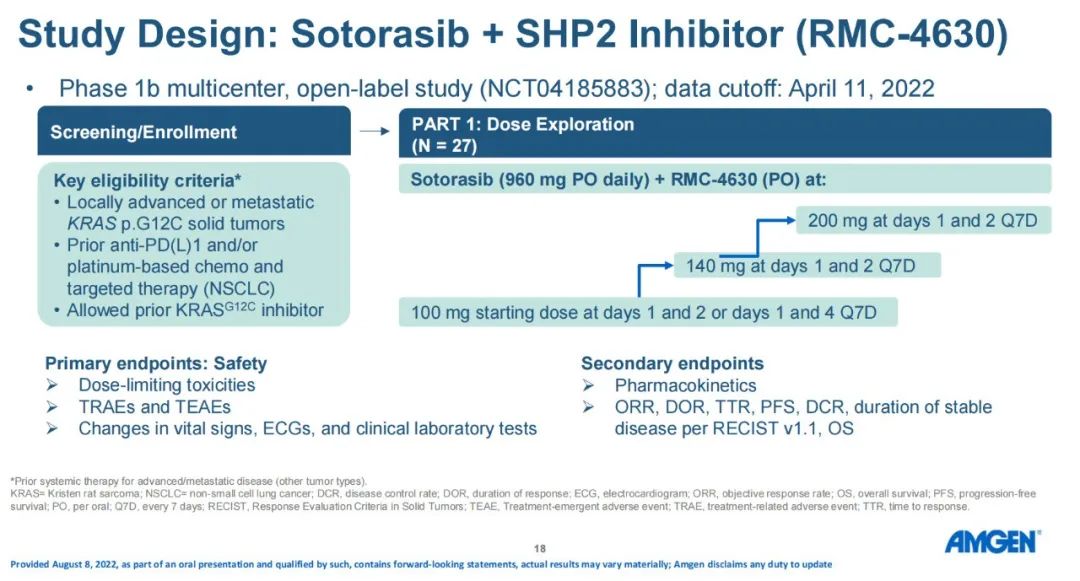
研究方法:这项多中心开放标签研究招募了局部晚期或转移性KRAS g12c突变的实体肿瘤患者。在NSCLC队列中,患者之前必须接受PD-1/PD-L1抑制剂和/或铂基化疗和靶向治疗。所有队列均允许使用过KRAS G12C抑制剂治疗。主要终点是安全性/耐受性;次要终点是客观缓解率(ORR)和药代动力学(PK)。
研究结果:数据截止日期为2022年4月11日,患者既往治疗的中位数为3。在NCLC患者中,10例(91%)接受过PD-1/PD-L1抑制剂治疗,5例(45%)接受过KRAS G12C抑制剂治疗。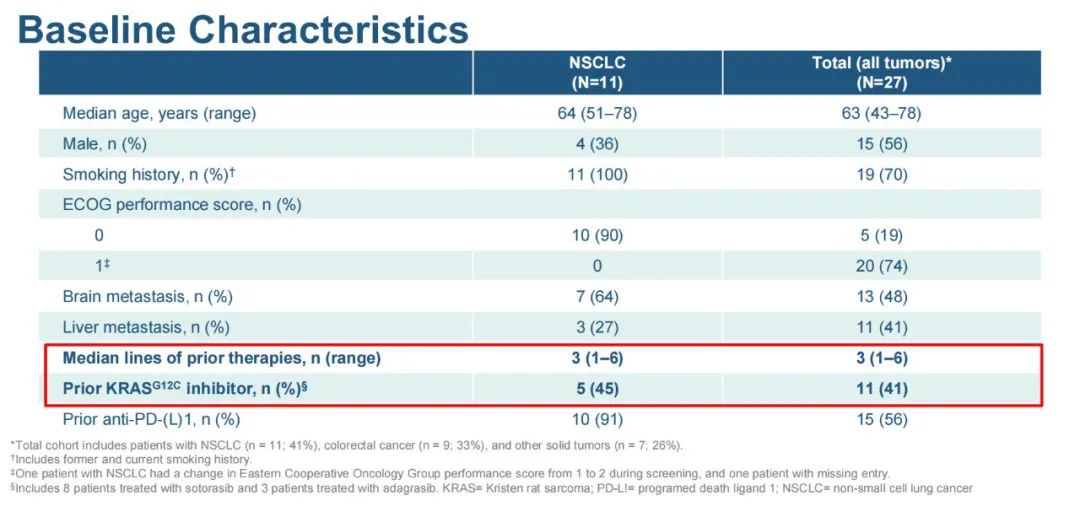
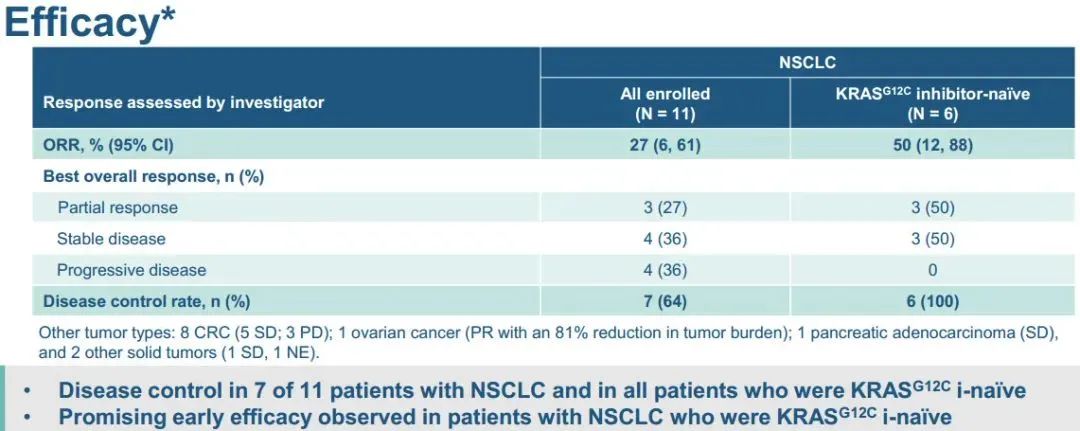
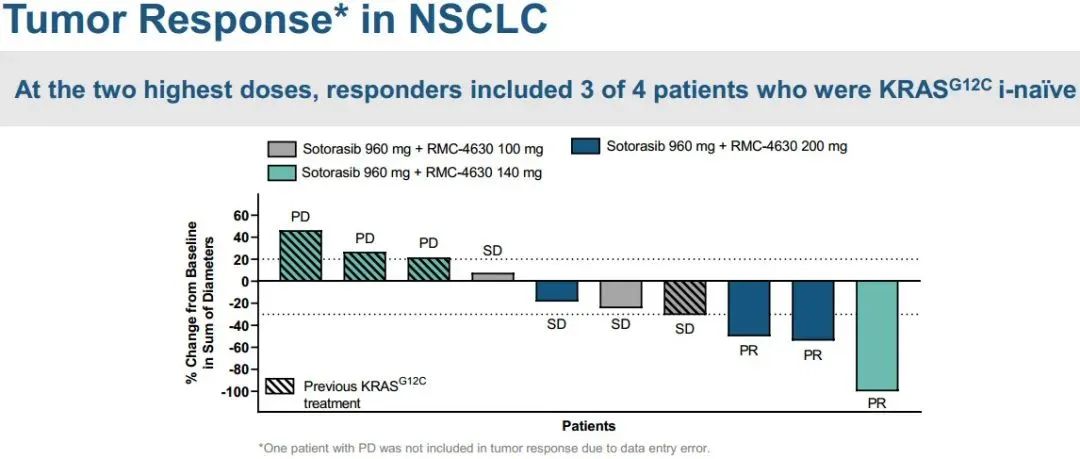
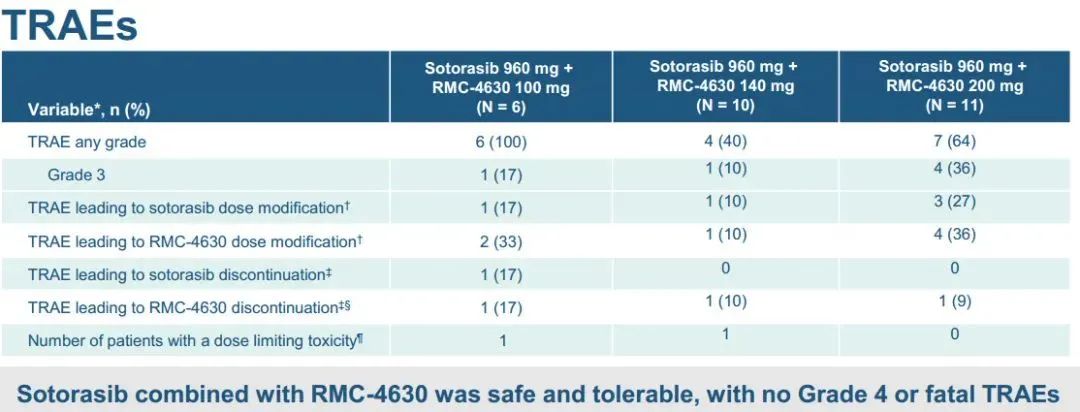
1)在所有接受联合治疗的患者中,3例出现部分缓解(PRs;27%),4例病情稳定(SD;36%),4例疾病进展(PD;36%)。2)在KRAS G12C抑制剂初治人群中,报告了3例PRs(50%)和3例SD(50%),即DCR为100%。3)联合方案的中位持续时间为3.1个月。在数据截止时,3例PR患者中有2例持续缓解,DOR分别超过9.8个月和6.9个月。此外,1例既往sotorasib治疗进展的患者在5.5个月时肿瘤缩小超过30%,6.9个月时出现PD。4)sotorasib和RMC-4630之间没有临床意义的药物-药物相互作用,sotorasib和RMC-4630的平均暴露量与单药治疗研究中观察到的分布一致。值得注意的是,在RMC-4630两个最高剂量组,4名患者中有3名应答者是KRAS G12C抑制剂初治患者。安全性:剂量递增完成时未发生任何≥4级治疗相关的不良事件(TRAEs)。63%的患者出现任何级别的TRAEs;最常见的是外周和局部水肿(30%),腹泻(26%),口干(11%)和疲劳(11%)。6例(22%)发生3级TRAEs ,其中腹泻2例,腹水、谷草转氨酶(AST)升高、结肠炎、呼吸困难、高血压、胸腔积液各1例。NTRK基因融合是包括肺癌在内的多种类型肿瘤的致癌驱动因子。拉罗替尼是一种高度选择性、具有中枢神经系统 (CNS) 活性的原肌球蛋白受体激酶 (TRK) 抑制剂。截至2020年7月【数据来自NCT02576431, NCT02122913两项临床试验】,拉罗替尼在15例TRK融合肺癌患者中经独立审查委员会 (IRC) 评估的客观缓解率 (ORR) 为 87% (Drilon et al, JCO Precis Oncol 2021)。 本次会议更新了这些患者的长期随访数据以及扩展数据。截止日期2020年07月,评价了15例晚期肺癌患者,IRC评估的ORR为87%,INV评估的ORR为73%。额外随访一年后,IRC评估的ORR仍为87%,在本分析中称为队列1。另外,在截至2021年7月,共纳入26例NTRK基因融合肺癌患者(定义为队列2,包括队列1的15例加上另外11例患者)。队列1、2疗效反应

队列2生存数据
研究结果:1)队列2的23例可评价患者中,有2例完全缓解、17例部分缓解(PR)和4例疾病稳定(SD)患者。无患者出现疾病进展。所有26例患者均未达到中位PFS和DOR,而中位OS为40.7个月(95%CI,19.4-无法估计[NE])。24个月DOR率为72%。DOR的中位随访时间为12.9个月,而PFS为14.6个月,OS为12.9个月。2)10例可评价患者有基线中枢神经系统转移。他们的ORR为80%(95%CI,44%-97%),包括8例PR和2例SD。对于这些患者,中位DOR为9.5个月(95%CI,3.6-NE),而中位PFS为9.9个月(95%CI,5.3-NE),中位OS为19.4个月(95%CI,7.6-NE)。3)另外,队列1的15例肺癌患者,经24个月随访后,mDoR未达到,35.8个月随访后,mPFS和mOS分别为33个月和40.7个月。安全性:队列1没有新的安全性信号,该组之前主要报告1级和2级AE。队列2也主要报告1级和2级治疗相关AE。最常见的治疗相关AE包括丙氨酸氨基转移酶和天冬氨酸氨基转移酶(AST)水平升高(分别为38.5%和30.8%),其次是肌痛(26.9%)和便秘(23.1%)。与larotrectinib相关的最常见≥3级AE为AST升高(7.7%)和体重升高(7.7%)。没有患者因AE中止治疗。TROPION-Lung02(NCT04526691)研究是一项Ib期、全球性、剂量递增和扩展研究,旨在评估两个剂量水平Dato-DXd(4mg/kg和6mg/kg)与帕博利珠单抗±铂类化疗(顺铂/卡铂)方案治疗初治或经治且无可靶向基因改变(如EGFR、ALK、ROS1、NTRK、BRAF、RET、MET或其他已知的可靶向基因改变)的晚期或转移性NSCLC患者的安全性和疗效。总共评估6个队列,每个队列约20例患者,队列扩展中的患者必须为未经治的晚期/转移性NSCLC。主要目标是评估耐受性和安全性。次要目标是评估疗效、药代动力学和抗药物抗体。
人滋养层细胞表面抗原-2(TROP-2)是由TACSTD基因家族编码的一种单次跨膜表面糖蛋白,又被称为肿瘤相关钙离子信号转导子2(TACSTD2)、表皮糖蛋白1(EGP-1)、胃肠肿瘤相关抗原(GA733-1)、表面标志物1(M1S1),TROP-2表达于几乎所有亚型的肺癌中,但高表达于腺癌(64%)和鳞状细胞癌(75%)。
Datopotamab deruxtecan(Dato-DXd)是一种靶向TROP-2的抗体药物偶联物(ADC),此前,TROPION-PanTumor01(NCT03401385)研究表明,Dato-DXd作为单药治疗复发/难治性晚期/转移性NSCLC患者6mg/kg剂量组的ORR为28%,DoR为10.5个月,显示出令人鼓舞的疗效和可控的安全性。此外,DXd ADC联合PD-1抑制剂的临床前肿瘤消退效果优于任何单药治疗。
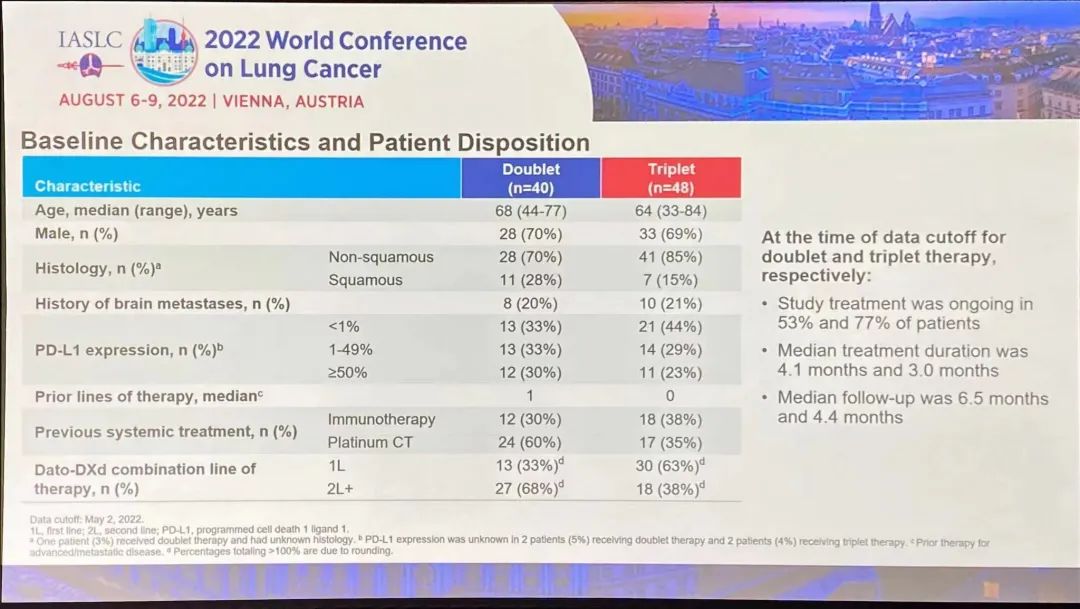
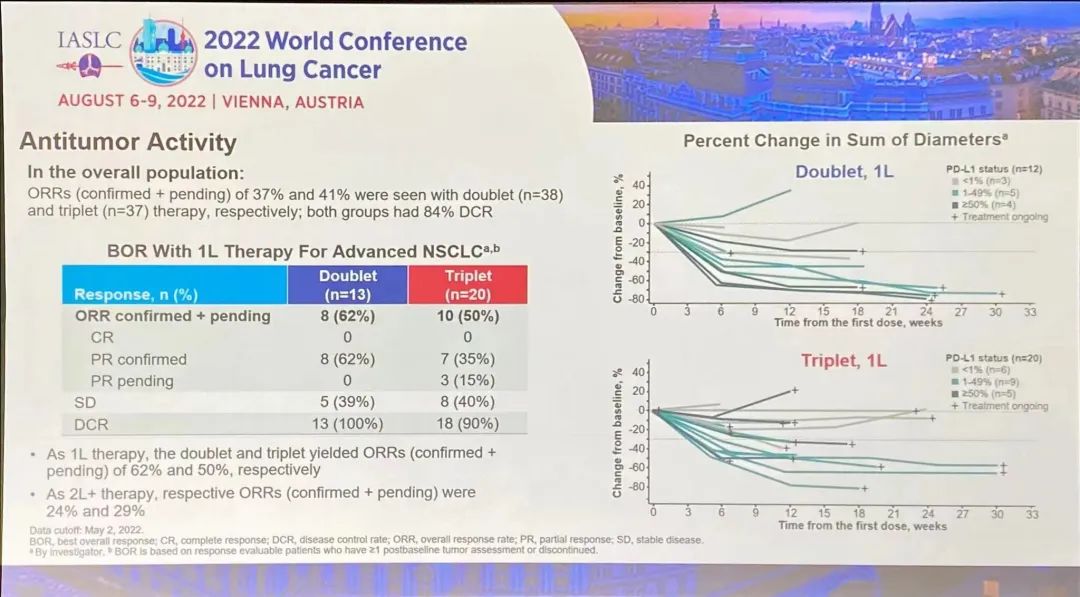
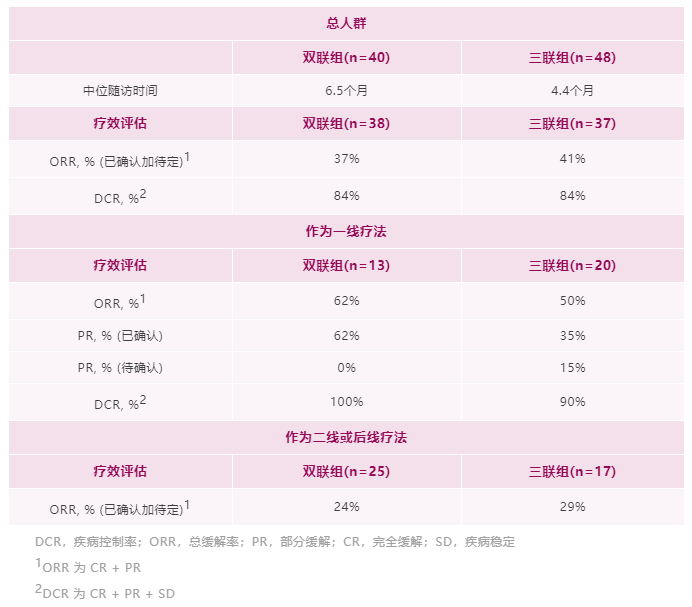
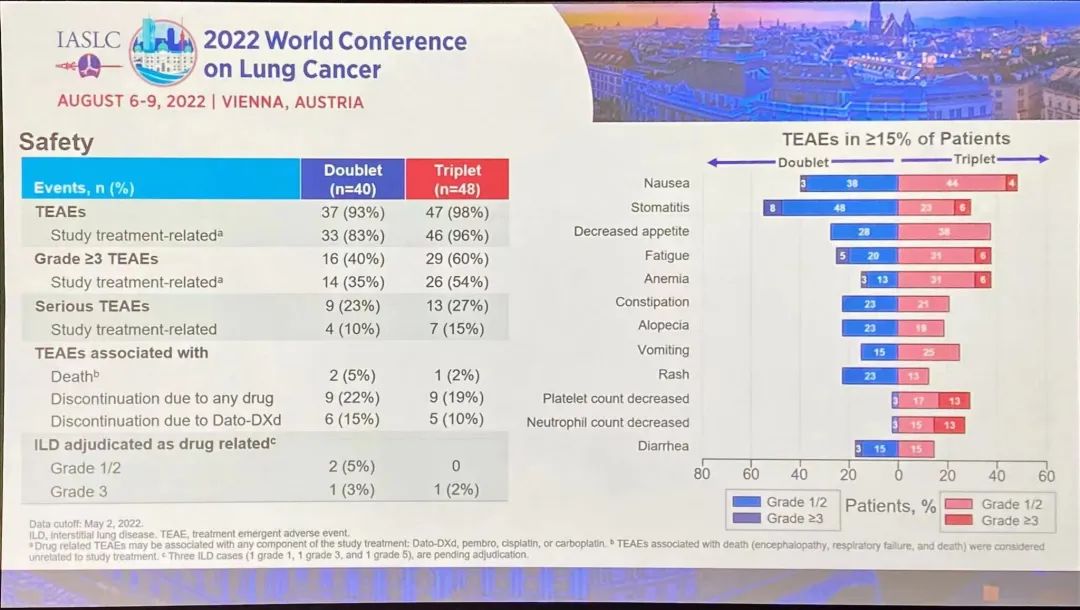
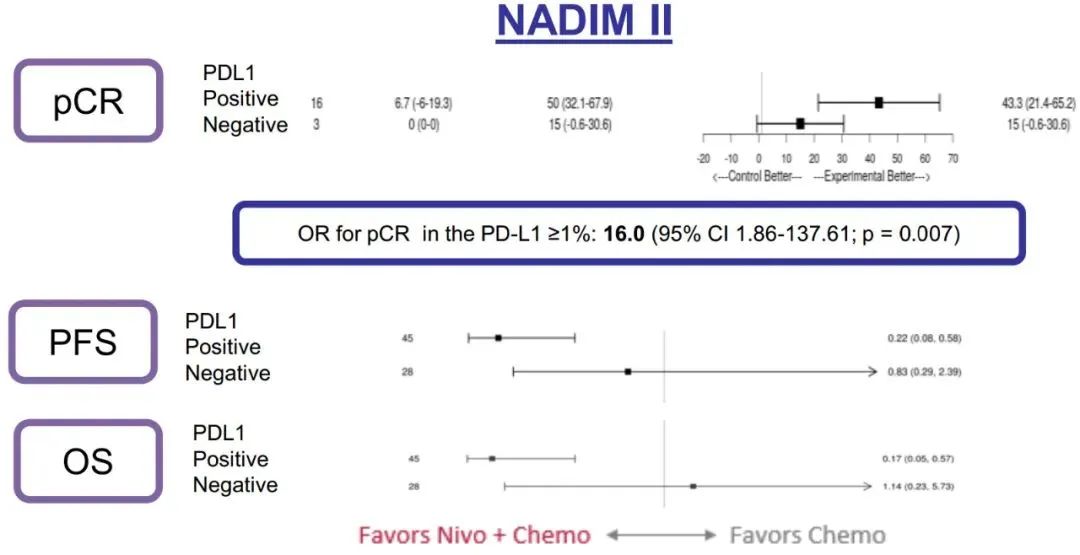
研究结果:TROPION-Lung02 研究中,双联组【datopotamab deruxtecan+帕博利珠单抗】的患者,平均接受过1线的先前治疗,包括铂类化疗(60%)和免疫治疗(30%)。在三联组【datopotamab deruxtecan+帕博利珠单抗+铂类化疗】中,患者之前接受过铂类化疗(35%)和免疫治疗(38%)。在双联组和三联组中,使用datopotamab deruxtecan 联合治疗方案作为一线治疗的患者占比分别为 33% 和 63%。截至 2022 年 5 月 2 日数据截止,分别有 53% 和 77% 的患者继续接受双联疗法和三联疗法。1)对于无可靶向驱动基因组改变的初治或经治的晚期或转移性 NSCLC 患者,治疗方案在总人群中显示出了良好总缓解率 (ORR),双联疗法ORR为37%(中位随访时间为 6.5 个月),接受三联疗法的患者的 ORR 为 41%(中位随访时间为 4.4 个月)。在包括一线和二线治疗的总人群中,双药和三药联合治疗的疾病控制率 (DCR) 为 84%。2)在未接受过治疗的患者中, ORR分别为62%(接受双药治疗的 13 名患者中有 8 名)和 50%(接受三药治疗的 20 名患者中有 10 名)。在接受双药治疗的患者中观察到 8 名患者出现部分缓解 (PR),在接受三药治疗的患者中观察到 10 名患者出现 PR(3名待确认)。双药治疗的 DCR 为 100%,三药治疗的 DCR 为 90%。3)安全性:Datopotamab deruxtecan 的联合治疗方案显示出了可耐受的安全性,这支持了正在进行的研究评估。在双联组和三联组中,分别有 40% 和 60% 的患者发生 了3 级或以上的治疗期间出现的不良事件 (TEAE)。在双联组和三联组中最常见的任何级别的 TEAE 分别是口腔炎(56% 和 29%)、恶心(41% 和 48%)、食欲下降(28% 和 38%)、疲劳(25% 和 37%) 和贫血 (16% 和 37%)。在两个队列中,有4例间质性肺病(ILD)事件被独立判定委员会确定为与药物相关;两例被判定为 1/2 级事件,两例被判定为 3 级事件。没有 4 级或 5 级 ILD 事件被判定为与药物相关。在数据截止时,有3例潜在的 ILD 事件等待判定。发生的 3 例死亡(2 例在双联组中,1 例在三联组中)均未确定为与药物相关。因不良事件而中断治疗的患者数量低于22%,13% 的患者发生了 datopotamab deruxtecan 剂量中断。小 结:Dato-DXd联合帕博利珠单抗±铂类在未经治复发/难治性NSCLC患者中,表现出了显著的抗肿瘤活性和可耐受的安全性特征。据了解,这是首次公开报道的TROP-2 ADC联合免疫检查点抑制剂和铂类药物治疗NSCLC的临床经验。作为一款新型TROP-2导向的ADC药物,Dato-DXd的研究数据让大家看到了这种药物的潜力,期待后续能有更好的临床试验结果。NADIM II(NCT03838159)是一项开放标签、随机、双臂、II期多中心临床研究(NADIM试验的后续研究)。可切除IIIA-B期NSCLC、ECOG PS 0-1 且EGFR/ALK阴性患者随机分配接受“化疗±免疫”新辅助治疗,序贯手术治疗。经病理学评估证实为 R0 切除的患者在术后第3-8 周内开始给予辅助纳武利尤单抗治疗(480 mg Q4W),并持续 6个月。主要终点为pCR。次要终点包括PFS、OS和生物标志物分析。今年ASCO大会上,NADIM II研究首次公布结果,与化疗相比,新辅助纳武利尤单抗+化疗明显改善可切除IIIA-B期NSCLC患者主要终点(pCR率,36.8% vs 6.9%,RR=7.88)。本次会议研究者公布了PFS和OS结果。
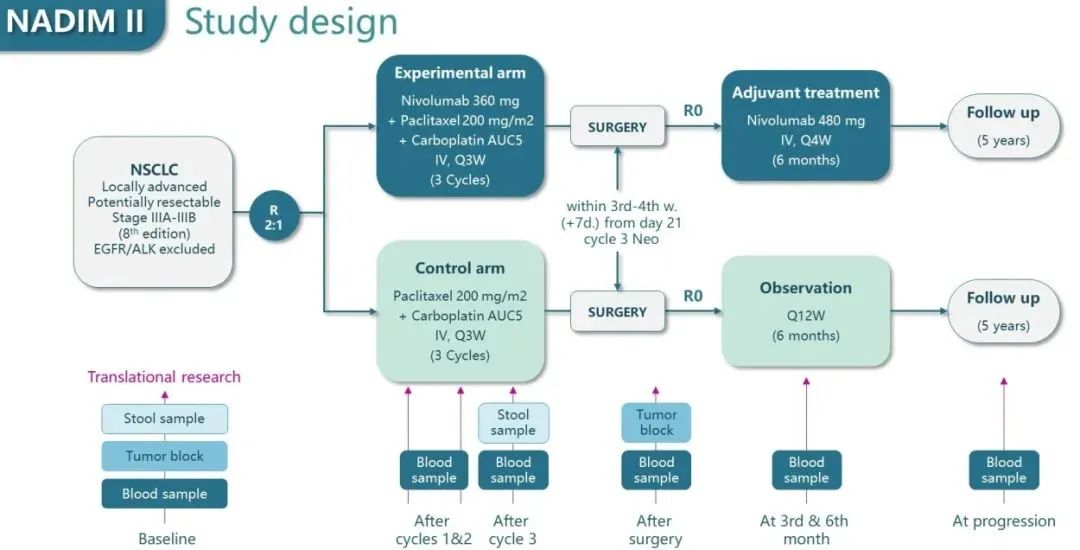
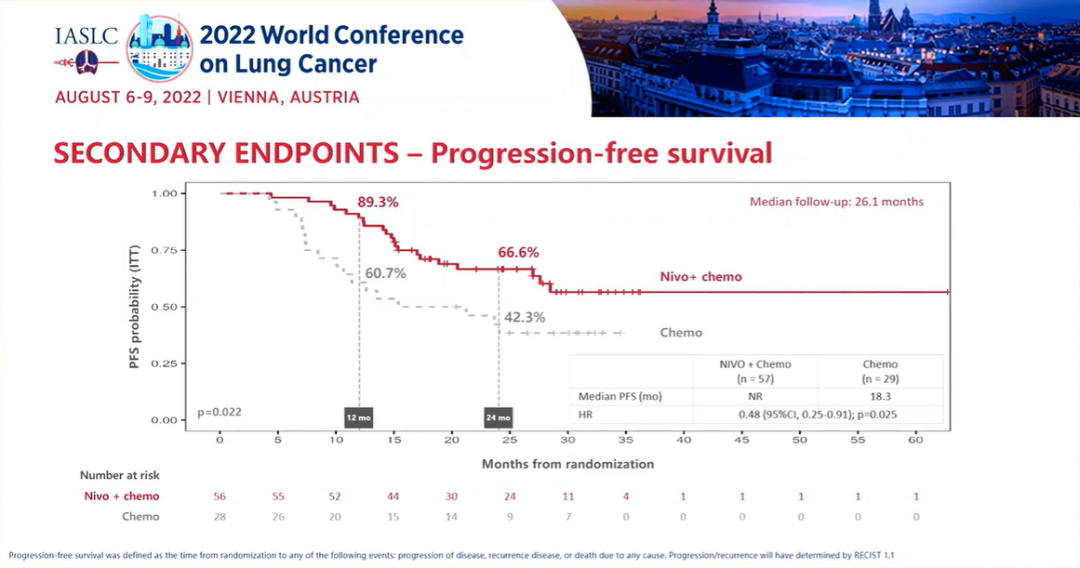
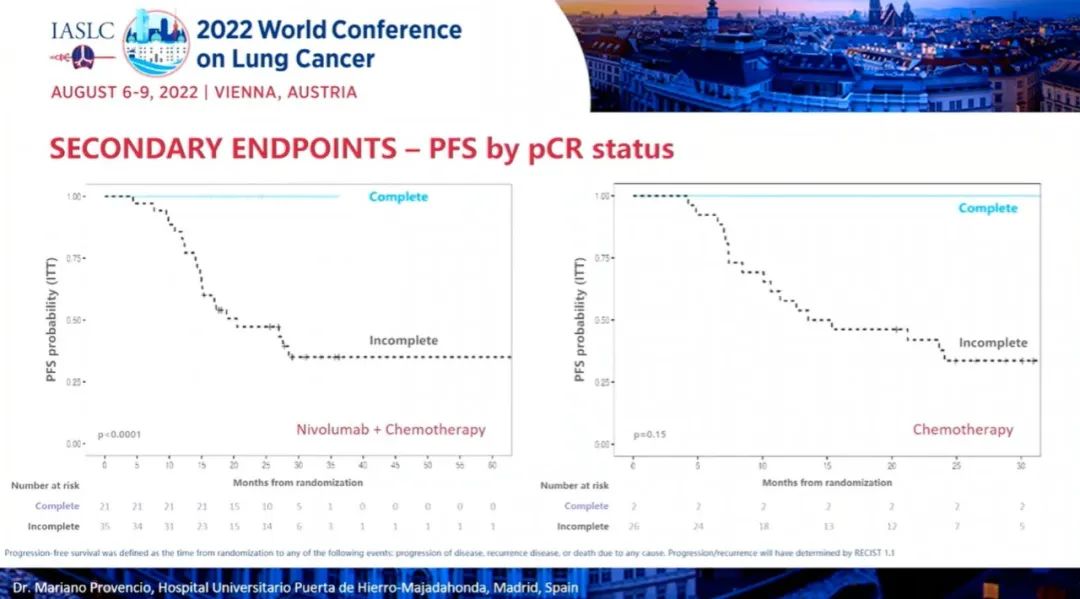
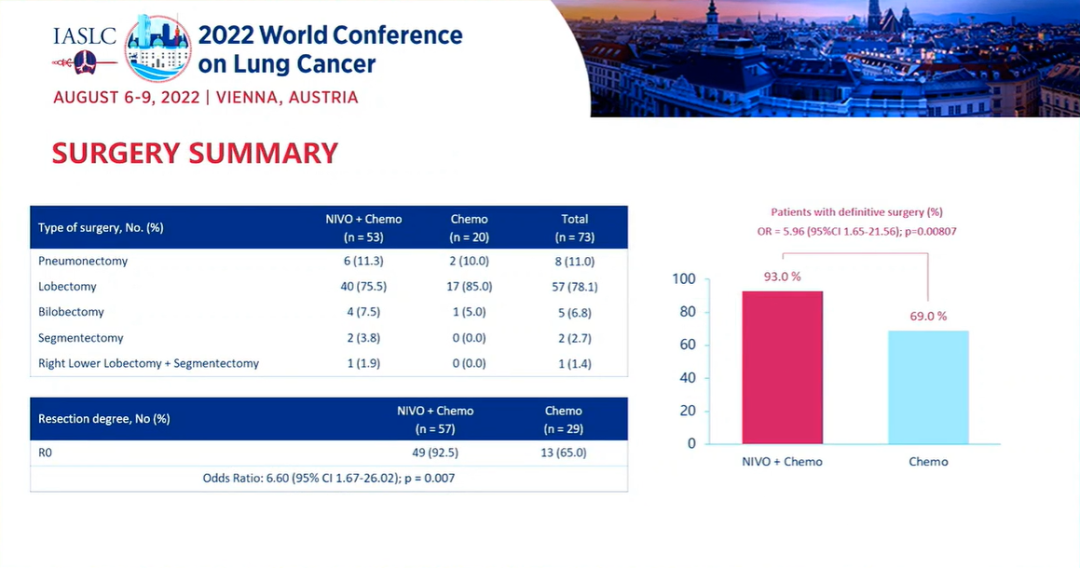
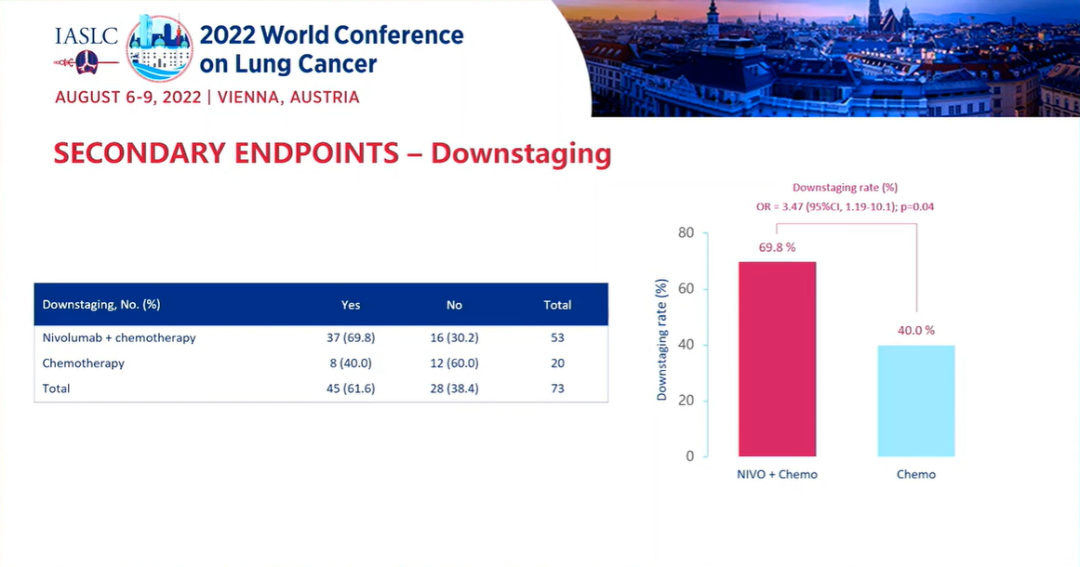
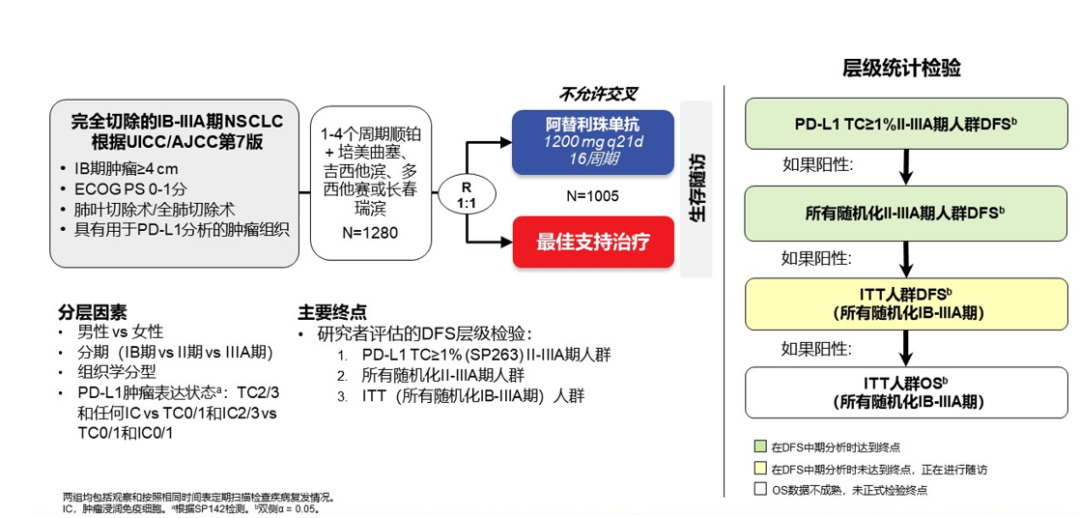
研究结果:中位随访时间为26.1个月。2021年3月数据截止时,免疫联合组、化疗组患者的24个月PFS率分别为66.6% vs 42.3%,中位PFS分别为 未达到 vs 18.3个月(HR=0.48; 95%CI:0.25-0.91; P=0.025)。免疫联合组、化疗组患者的24个月OS率分别为84.7% vs 63.4%,两组均未达到中位OS(HR=0.40; 95%CI: 0.17-0.93; P=0.034)。另外,PFS和OS数据显示,出现pCR缓解的患者均未出现疾病进展或死亡。免疫联合组、化疗组患者接受确定性手术的比例是93.0% vs 69.0%(OR=5.96, 95%CI: 1.65-21.56,P=0.00807);手术降期率分别为69.8% vs 40.0%(OR=3.47, 95%CI: 1.19-10.1,P=0.04)。研究结果证实,免疫联合化疗可为可切除 IIIA-B 期 NSCLC 患者带来生存获益。在PD-L1 表达阳性和达到 pCR的患者中,生存获益尤其明显。NADIM ll研究是首个新辅助免疫联合治疗在可切除IIIA~B期NSCLC患者中显示出OS改善的临床试验。IMpower010研究:是一项多中心、开放标签、III期随机对照研究,研究在22个国家和地区的227个地点进行。共有1280名IB期(肿瘤≥4 cm)至IIIA期NSCLC患者在完全切除后入组,通过区组随机化的方法(4 个区组大小)将接受辅助铂类化疗(1到4个周期)后的患者随机分配(1:1)接受辅助阿替利珠单抗(每 21 天 1200 mg;16 个周期或 1 年)或最佳支持治疗(观察和定期监测疾病复发)。
研究主要终点为研究者评估的DFS,次要终点为总生存(OS),进行分层分析:首先分析PD-L1 TC≥1%(SP263)的II-IIIA期NSCLC患者亚组,然后评估所有随机II-IIIA患者的DFS,然后在分析所有意向治疗(ITT)人群(IB-IIIA患者)的DFS和最终OS。疗效评估是基于随机的患者。安全性在可评估人群中(接受≥1剂阿替利珠单抗或BSC组接受≥1次基线后安全性评估的患者)进行。
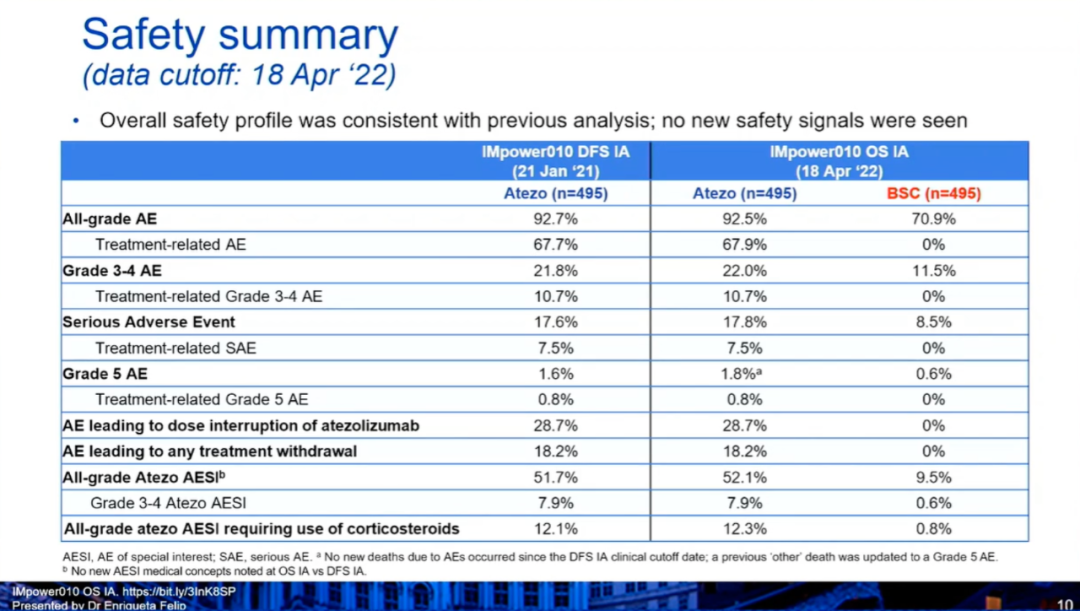
之前第一次DFS期中分析(中位随访32.2个月后),与最佳支持治疗相比,阿替利珠单抗显著延长了无病生存。PD-L1表达≥1%的II-IIIA期患者:疾病进展或死亡风险降低34%(分层HR 0.66),3年无病生存率更高(60% vs 48%)。所有II-IIIA期患者:疾病进展或死亡风险降低21%(分层HR 0.79),3年无病生存率更高(56% vs 49%)。本次会议公布了第一个预先指定的OS期中分析和安全性分析,中位随访期为45.3个月,数据截止日期为2022年4月18日。
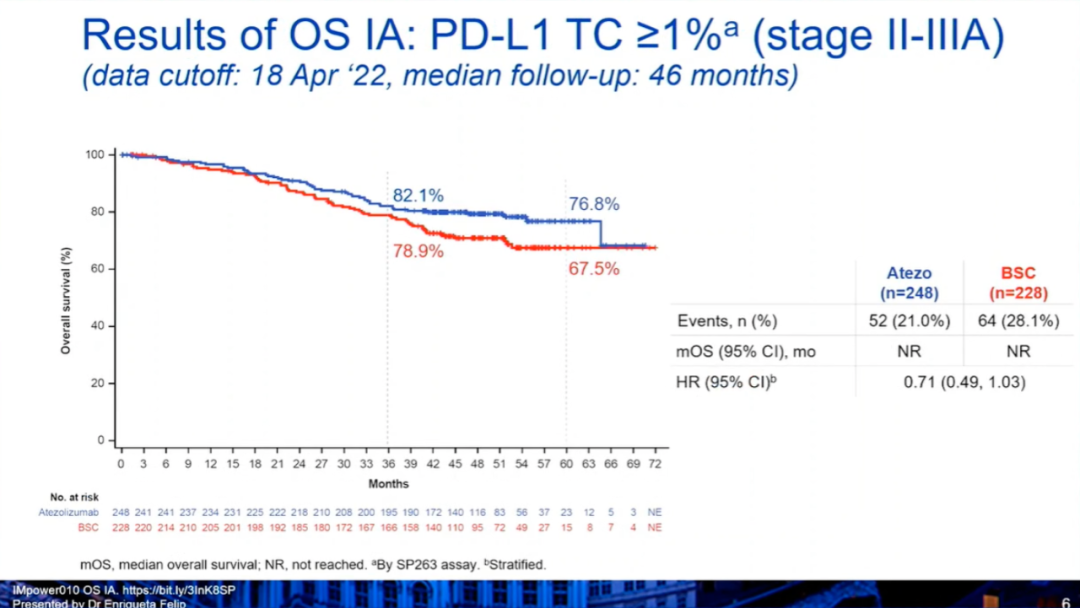
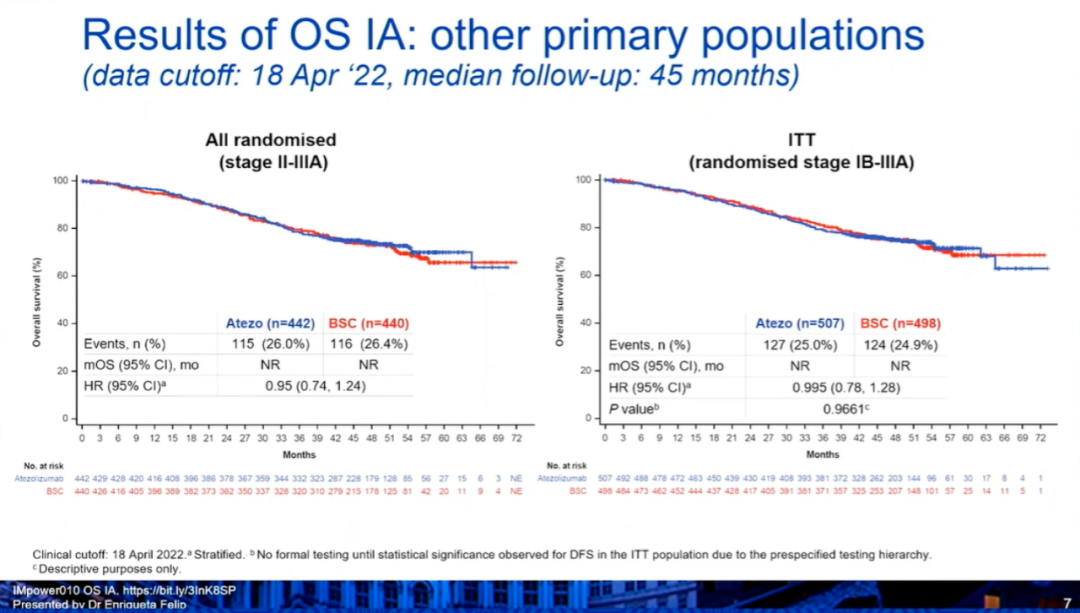
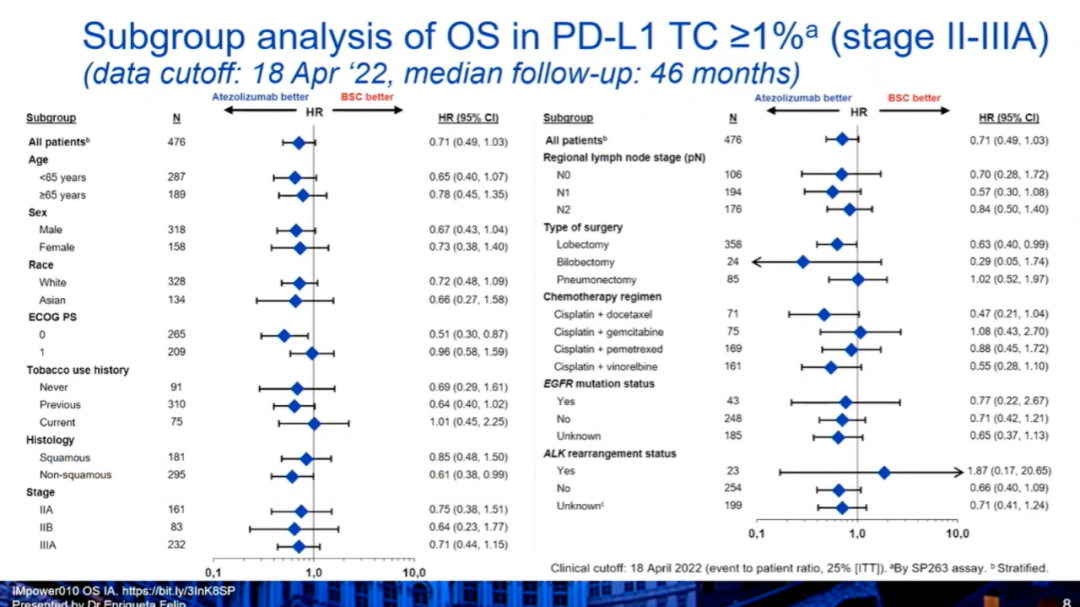
研究结果:OS期中分析显示,1)在PD-L1≥1%的II~IIIA期人群中,阿替利珠单抗组和BSC组的中位OS均未达到(HR:0.71,0.49-1.03),36个月、60个月的OS率分别为82.1% vs 78.9%,76.8% vs 67.5%。2)在所有随机化人群中(II~IIIA期),阿替利珠单抗组和BSC组的中位OS均未达到(HR:0.95,0.74-1.24)。3)在所有意向治疗(ITT)人群(IB-IIIA患者)中,阿替利珠单抗组和BSC组的中位OS均未达到(HR:0.995,0.78-1.28,P=0.9661)。
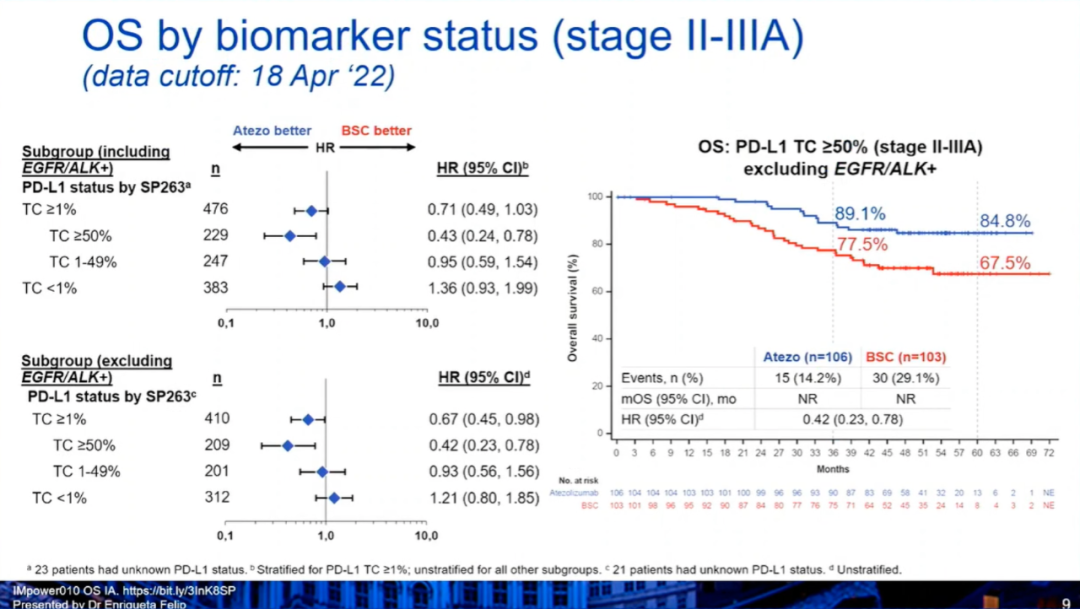
4)此外,在PD-L1≥50%(II~IIIA期,排除EGFR/ALK+)人群中,阿替利珠单抗组和BSC组的中位OS均未达到(HR:0.42,0.23-0.78),36个月、60个月的OS率分别为89.1% vs 77.5%,84.8% vs 67.5%。与2021年1月21日时的评估结果相似,阿替利珠单抗组治疗相关AE的发生率为67.9%,治疗相关3~4级AE的发生率为10.7%,治疗相关5级AE的发生率为0.8%。小 结:在第一次预先指定的OS期中分析中,在PD-L1≥1%的II~IIIA期患者中,阿替利珠单抗组可以看到OS的获益趋势。但是在所有随机化人群和ITT人群中,没有显示出OS的获益趋势。安全性数据与之前相似,没有出现新的安全性信号。期待IMpower010研究的最终DFS分析以及后续的OS分析。GEMSTONE-301研究:这是一项随机、双盲、安慰剂对照、III期临床试验,评估了舒格利单抗或安慰剂作为巩固治疗在含铂方案同步或序贯放化疗后未出现疾病进展的局部晚期、不可切除、III 期 NSCLC 患者中的疗效。在中国50家医院或学术研究中心进行。符合条件的患者被随机分配(2:1)至舒格利单抗组或安慰剂组。患者每3周接受一次舒格利单抗1200 mg 或匹配的安慰剂作为巩固治疗,持续时间长达 24 个月。分层因素为ECOG PS评分、既往放化疗史和总放疗剂量。
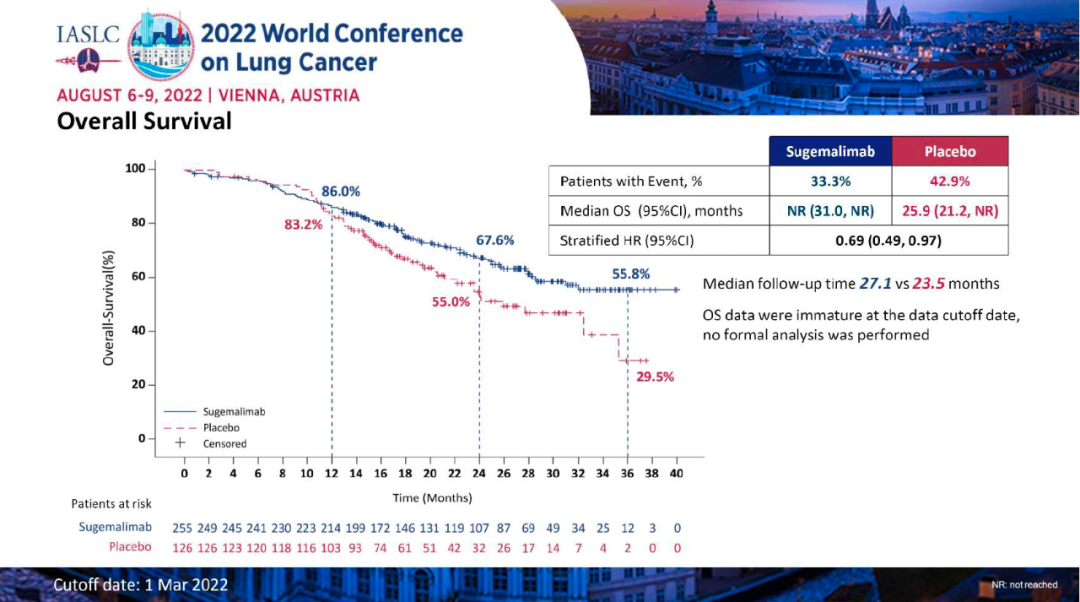
在中期分析的数据截止时,舒格利单抗组和安慰剂组分别有123例(48%)和74例(59%)出现疾病进展或死亡。舒格利单抗组和安慰剂组的PFS分别为9.0个月 vs 5.8个月(分层风险比 [HR] 0.64, 95%CI 0.48 –0.85;p=0·0026 )。两组的12个月无进展生存率为 45·4%和25·6%。在大多数亚组中,舒格利单抗相比于安慰剂相比均可见PFS获益。在数据截止时,OS数据仍不成熟。
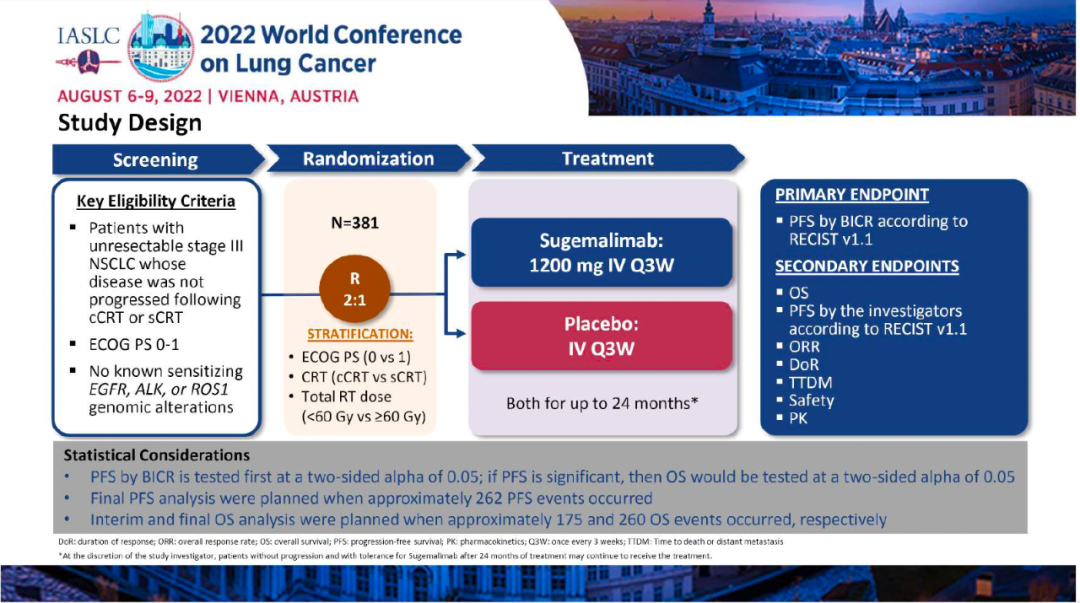
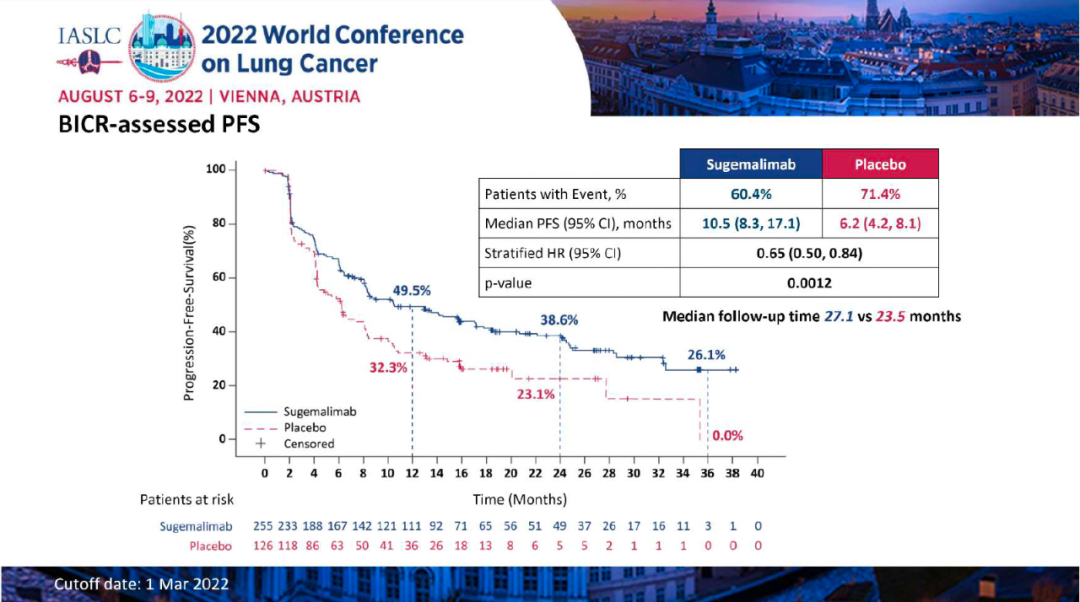
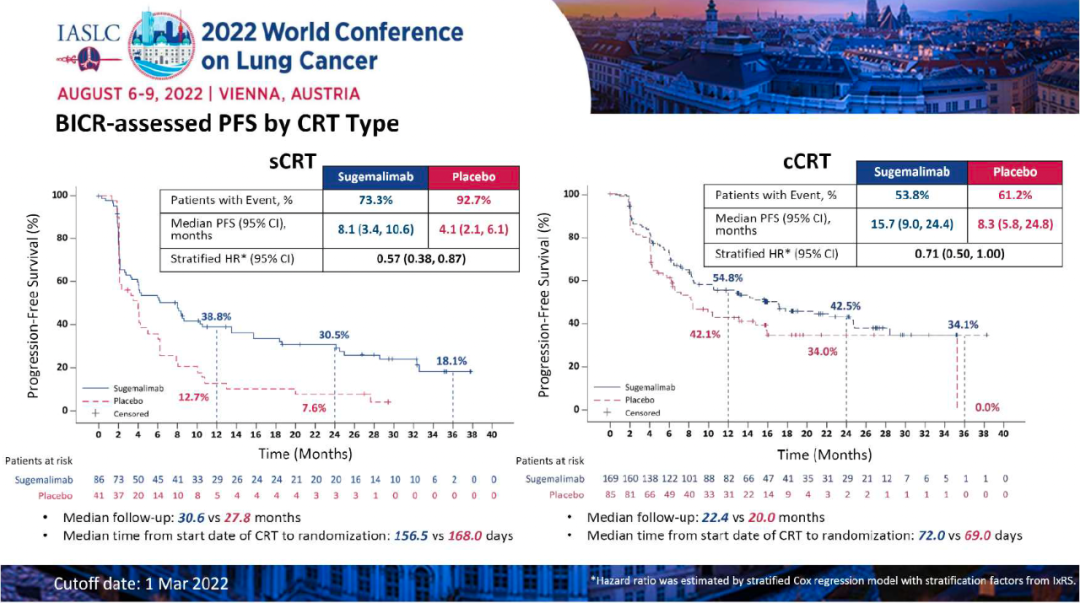
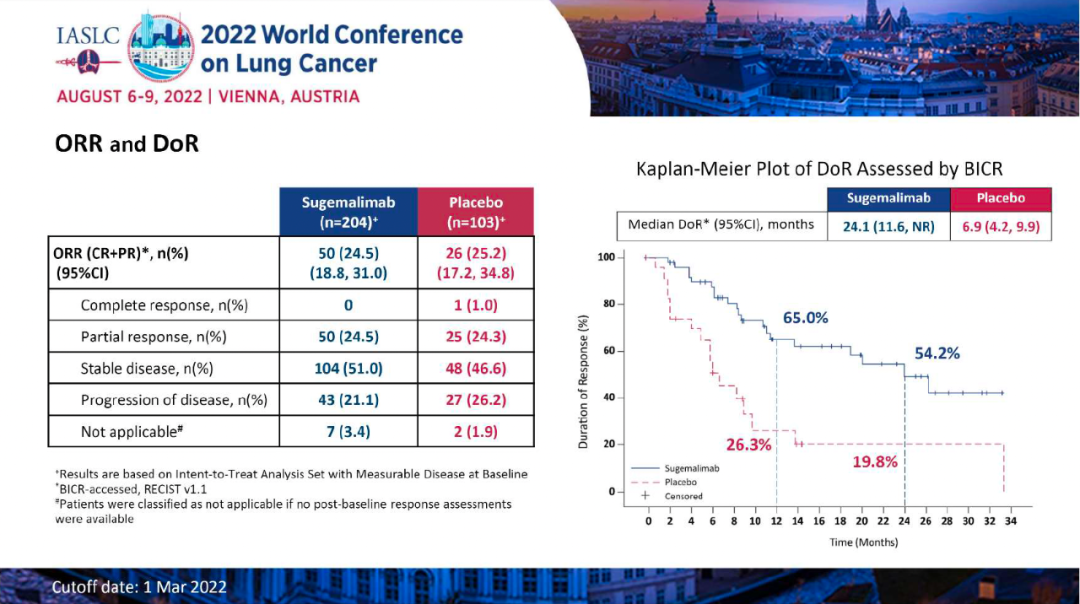
研究结果:本次WCLC会议报告了最终PFS分析的数据,至数据截止日期(2022年3月1日),1)舒格利单抗组和安慰剂组的中位随访时间分别为27.1个月和23.5个月,中位PFS分别为10.5 vs 6.2个月(HR:0.65,P=0.0012);其中接受序贯放化疗的患者,分别为8.1 vs 4.1个月(HR:0.57);接受同步放化疗的患者,分别为15.7 vs 8.3个月(HR:0.71)。2)在数据截止时,OS数据仍不成熟。初步数据显示,舒格利单抗组和安慰剂组的中位OS分别为NR vs 25.9个月(HR:0.69)。其中接受序贯放化疗的患者,分别为:未达到 vs 24.1个月(HR=0.60);接受同步放化疗的患者,分别为:未达到 vs 32.4个月(HR=0.75)。3)舒格利单抗组和安慰剂组的ORR分别为24.5% vs 25.2%,DoR分别为24.1 vs 6.9个月。
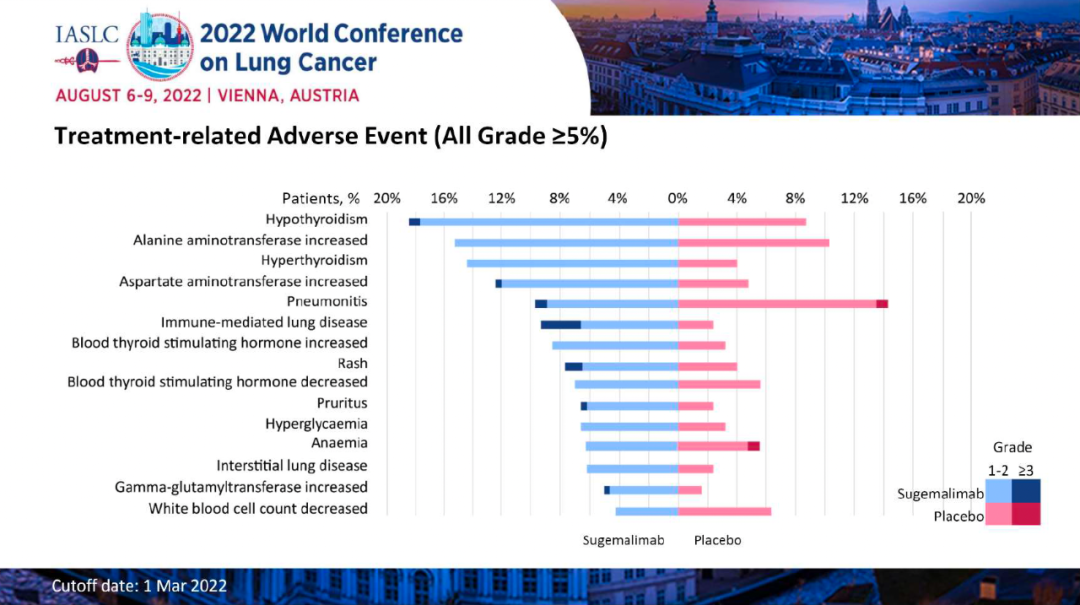
安全性:舒格利单抗组和安慰剂组的治疗相关治疗期不良事件(Treatment Emergent Adverse Event,TEAE)发生率分别为78.4%和64.3%,治疗相关3~5级TEAE发生率分别为11.4%和5.6%,因TEAE导致死亡分别有12例(4.7%)和3例(2.4%)。在无EGFR/ALK突变的晚期或转移性 NSCLC 患者中,与单独化疗相比,帕博利珠单抗作为单一疗法和联合化疗显著延长了OS和PFS。在临床试验设计中,完成35个周期(约2年)帕博利珠单抗治疗后疾病进展的患者可以接受帕博利珠单抗作为第二个疗程。本次会议对开始第二疗程帕博利珠单抗单药治疗的患者进行了探索性汇总分析报告。
研究方法:该分析汇总了 KEYNOTE-024、KEYNOTE-042 和 KEYNOTE-598 研究中接受帕博利珠单抗单药治疗(队列 1)以及 KEYNOTE-189 和 KEYNOTE-407 研究中接受帕博利珠单抗联合化疗(队列 2)治疗的晚期或转移性 NSCLC 患者。该分析中纳入的患者在完成 35 个周期的帕博利珠单抗(±化疗)后获得疾病稳定(SD)或更好缓解,或者因疾病完全缓解(CR)在35个周期前停止帕博利珠单抗治疗,在疾病进展(PD)后接受第二疗程的帕博利珠单抗治疗(最多 17 个周期)。在意向治疗(ITT)人群中分析疗效,在治疗人群中分析安全性。
在队列1共148 例完成 35 个周期帕博利珠单抗治疗并出现PD的患者中,58 例患者接受了第二疗程帕博利珠单抗治疗并被纳入本次分析。队列 2 纳入了16例(16/55)完成35 个周期帕博利珠单抗治疗的患者(作为帕博利珠单抗联合化疗的一部分),疗效评估为PD,并接受了第二疗程的帕博利珠单抗治疗。队列 1 中有18/58 例患者 (31%) 为鳞癌,队列 2 中有7/16 (44%)例;队列 1有 47/58 (81%) 例患者PD-L1 TPS ≥50%,队列 2 中有7/16 (44%) 例。
研究结果:从停止第一疗程帕博利珠单抗到开始第二疗程的中位时间在队列 1 中为 11.7 个月(范围3.8-35.6) ,在队列 2 中为 6.3 个月(范围0.9-18.2) 。在队列 1 中第二疗程的中位持续治疗时间为 8.3 个月,队列 2 中为 7.6 个月,估计分别有 62% 和 59% 的患者在 6 个月时继续第二个疗程。在第二疗程帕博利珠单抗治疗期间,研究者评估的ORR在队列 1 中为 19%,在队列 2 中为 6%。队列 1 中有 14 例患者 (24%) 、队列 2 中有4 例患者 (25%) 在第二个疗程期间或之后出现了治疗相关不良事件(TRAE),其中3-4 级TRAE队列1有 3 例 (5%) ,队列2有 1 例 (6%) ;未出现5级TRAE。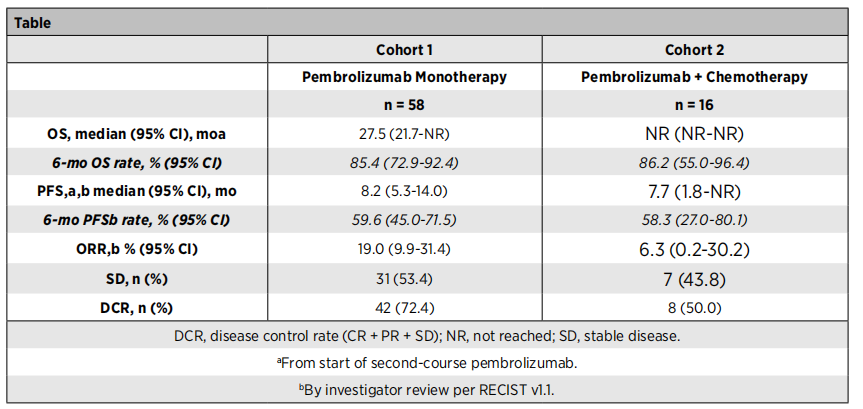
小结:在完成第一疗程帕博利珠单抗±化疗后出现 PD 的晚期或转移性 NSCLC 患者中第二疗程应用帕博利珠单抗单药治疗是可行的,显示出抗肿瘤活性以及具有临床意义的获益,以及可控的安全性。
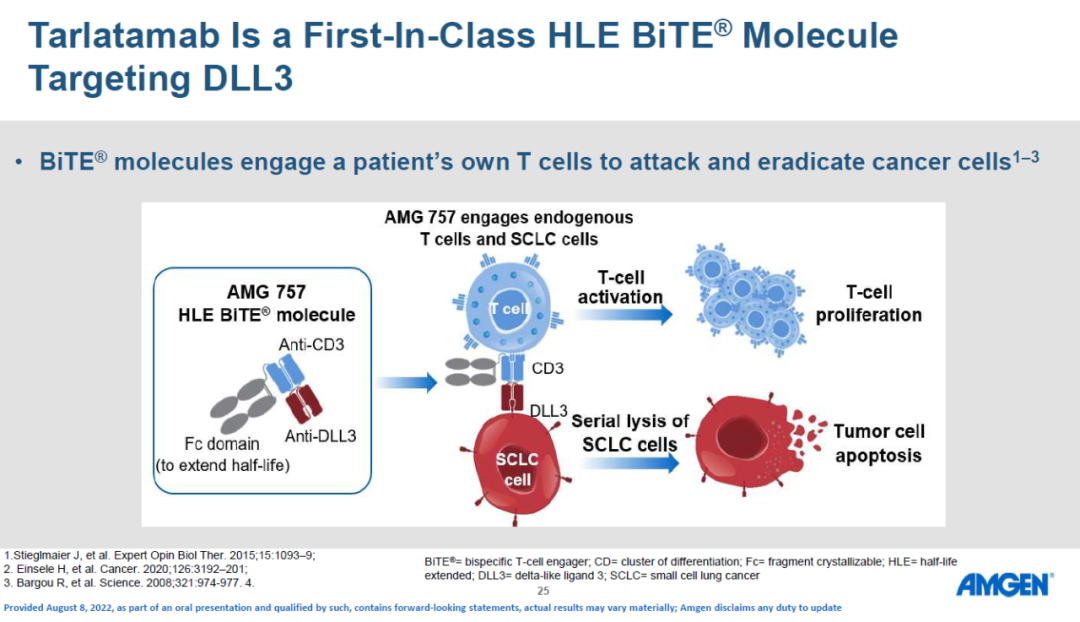
DLL3在大多数SCLC中过表达,Tarlatamab(AMG 757)是一种半衰期延长的双特异性T细胞衔接器(HLE BiTE®),可与DLL3和CD3结合,导致T细胞介导的肿瘤溶解。Tarlatamab治疗SCLC(NCT03319940)的中期I期剂量探索研究显示出初步的疗效证据,且具有可接受的安全性。研究人员首次报告了联合剂量探索和扩展队列的安全性、缓解率和生存数据。DeLLphi-300研究是Tarlatamab的首次人体试验,剂量探索范围为0.003-100.0 mg。主要终点是安全性和耐受性以及确定最大耐受剂量(MTD)和II期临床试验推荐剂量(RP2D),次要终点是PK参数和初步抗肿瘤活性。研究者基于改良RECIST v1.1评估药物的抗肿瘤活性。
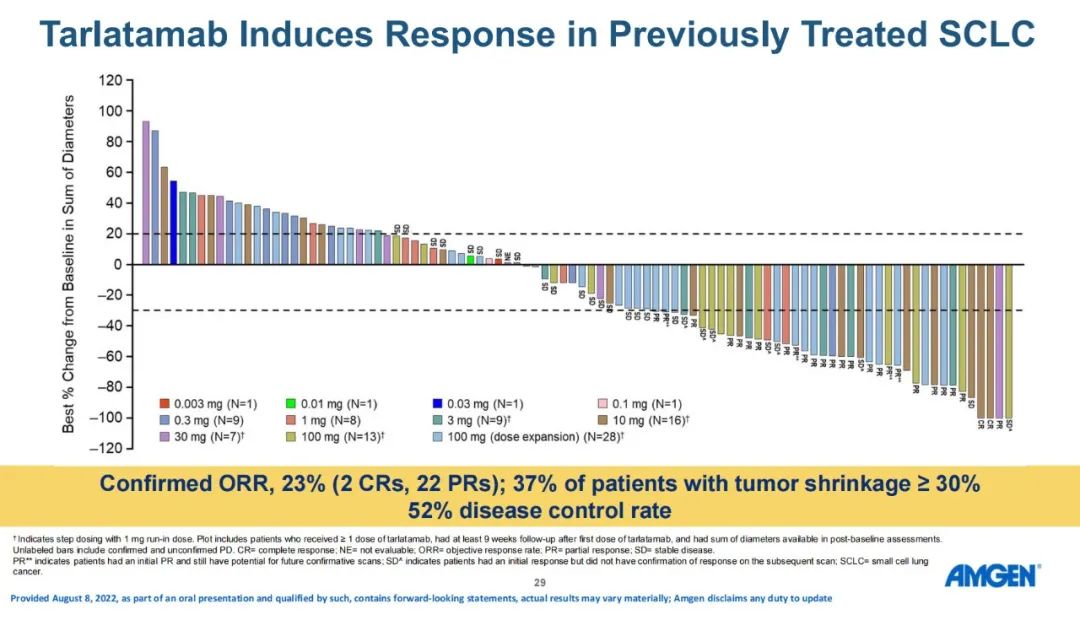
研究结果:研究纳入106例患者,接受过≥1线含铂化疗后疾病进展/复发,在剂量探索和扩展队列中接受了≥1剂Tarlatamab,中位随访时间为8.5个月。在中位前期治疗数为2的经治SCLC患者中,tarlatamab达到23%的确认ORR,和52%的疾病控制率。37%的患者肿瘤缩小超过30%,中位缓解持续时间为13.0个月,中位总生存期为13.2个月。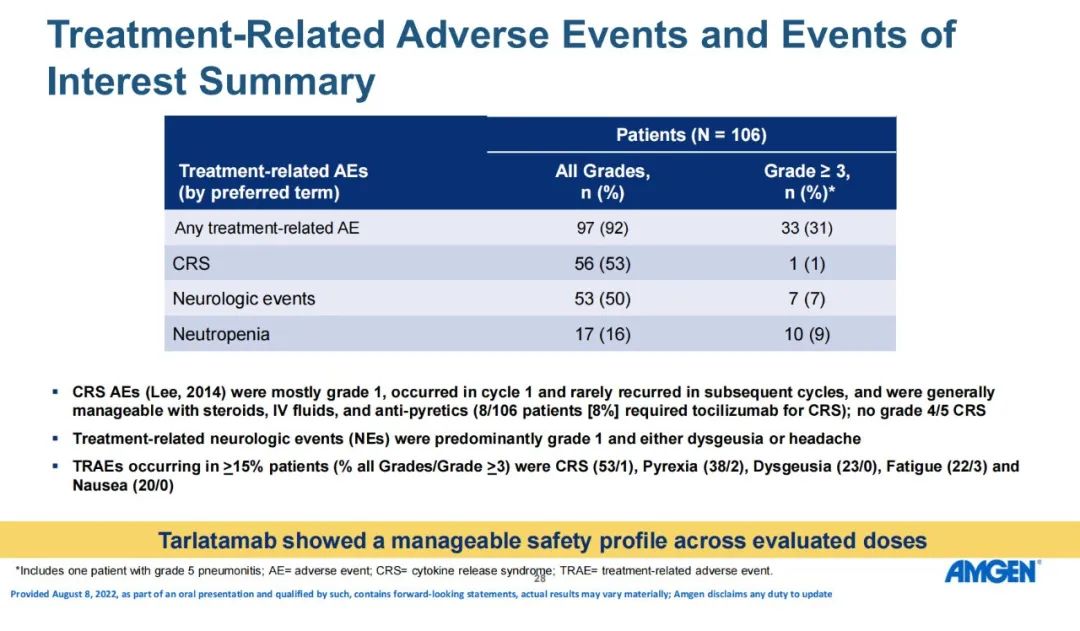 安全性
安全性
安全性方面,92%患者发生任何级别的治疗相关AEs(TRAEs),其中≥3级为31%。1例患者(1.0%)发生与治疗相关的3级CRS,7例患者(6.9%)发生治疗相关3级神经系统事件。3例患者(2.9%)因 TRAE(CRS、脑病和肺炎)而停用 tarlatamab。 小结:Tarlatamab具有预期的和可控的安全性,并在接受重度预处理确认缓解的SCLC人群中具有良好的应答持久性,表现出值得期待的疗效。PFS/OS与目前可用于复发性SCLC的其他疗法相比表现良好。基于这些结果,Tarlatamab 2L+治疗SCLC的II期研究DeLLphi-301(NCT05060016)正在进行。
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。
















































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)