
2022-11-18 来源 : 学术查
今天推荐的思路是中山大学第七附属医院发表在Cellular Oncology上题为“CDK7 blockade suppresses super‐enhancer‐associated oncogenes in bladder cancer”即对CDK7的阻断抑制膀胱癌中的超增强子相关的癌基因。

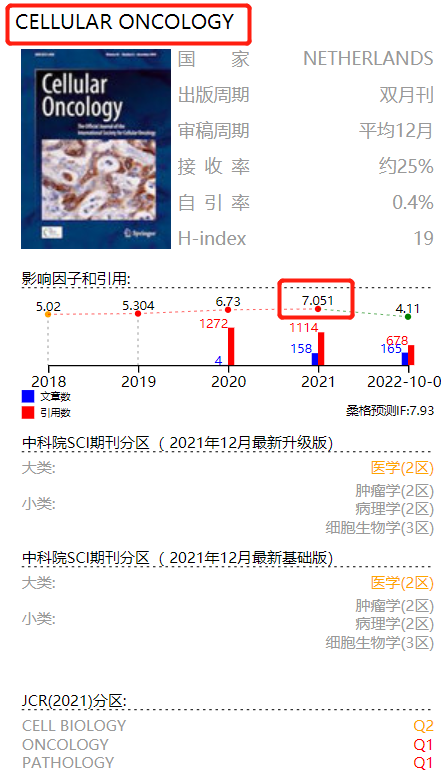
由基因改变引起的转录程序失调可能导致基因表达程序的显著变化,从而导致恶性细胞转化。靶向调节癌基因转录活性的超级增强子(SEs)已成为癌症治疗的一种有吸引力的策略。然而,迄今为止,膀胱癌 (BC) 中这一过程的分子机制仍有待阐明。作者发现 THZ1 作为一种有效的抑制剂,对 BC 细胞具有抑制活性。THZ1 和 DDIT4 抑制剂雷帕霉素联合治疗有效抑制了 BC 细胞的生长。
CDK7 是基因转录的一个特别重要的调节因子。为了评估 CDK7 在 BC 中的临床意义,作者评估了 GSE48276 数据集中 73 名 BC 患者的 CDK7 表达与总生存率之间的相关性。CDK7表达与BC患者预后呈负相关。随后 CRISPR/cas9 介导的 MGH-U3 细胞中 CDK7 敲低显示,细胞集落形成能力随着CDK7 蛋白表达下降而下降。表明强调CDK7 在 BC 细胞中的致癌作用。
前期研究已经发现 THZ1 与 CDK7 的活性位点对接,并且可以在 3-D 结构中观察到 THZ1 和 CDK7 在 Cys312 残基处的相互作用。这里,作者又发现 THZ1 显著抑制 10 种人 BC 细胞系的增殖,IC50值范围为 57 至 368 nM/L,表明 THZ1 可能是 BC 的有效抑制剂。
由于 5637 和 MGH-U3 是两种最敏感的细胞系,作者选择其进行体外研究,发现 THZ1 对 BC 细胞表现出显著的抗增殖作用。同时还发现 THZ1 导致 MGH-U3 和 5637 细胞大量凋亡。接下来作者还评估了 THZ1 处理对 BC 细胞周期进程的影响,发现用增加剂量的 THZ1 处理的 MGH-U3 和 5637 细胞在 G2/M 阶段表现出明显的细胞周期停滞。
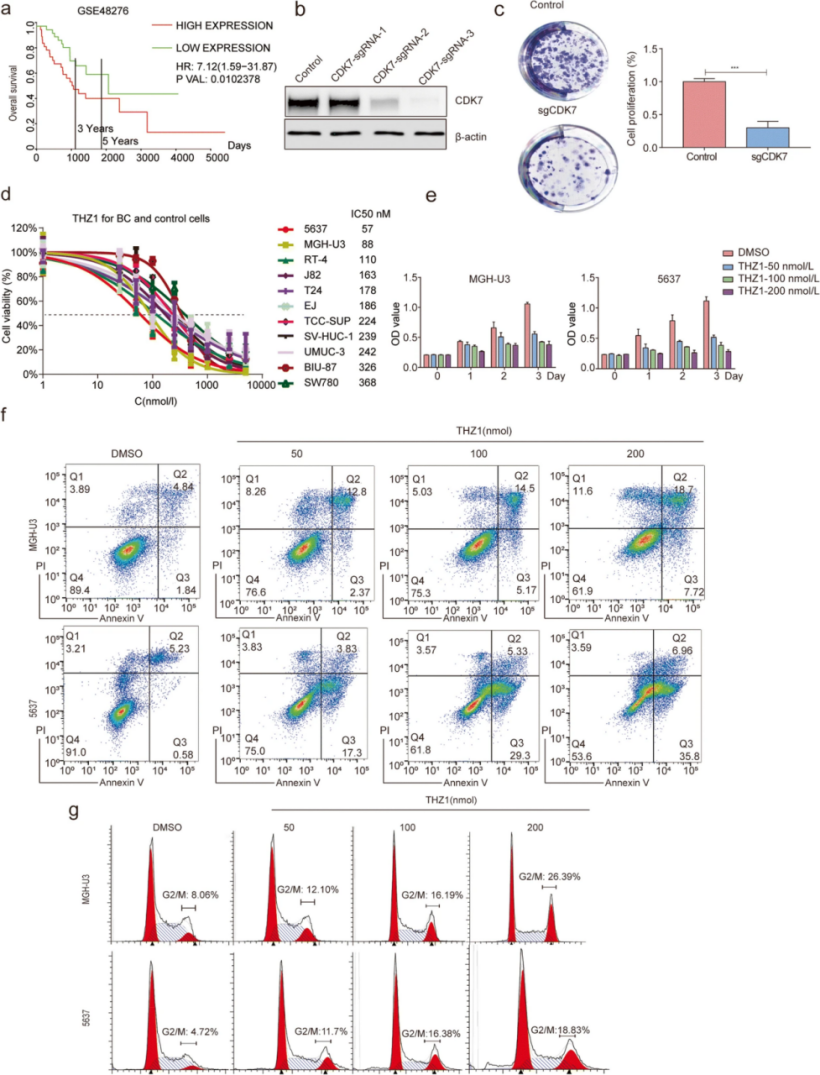
作者进一步研究 THZ1 对 BC 细胞的细胞毒性作用的潜在机制。据报道,CDK7 可能在转录调控中发挥双重作用 [21, 22]。已知 CDK7 参与转录起始相关丝氨酸 5 (S5) 和丝氨酸 7 (S7) 的磷酸化 [21, 23]。此外,CDK7 与 RNAPII C 末端结构域 (RNAPII CTD) 的延伸相关丝氨酸 2 (S2) 磷酸化的调节有关 [22, 24]。作者发现在 6 小时内逐渐增加 THZ1 剂量后,MGH-U3 和 5637 细胞中 RNAPII 的 S2、S5 和 S7 的磷酸化有类似的下降趋势。THZ1 可以选择性地抑制 RNAPII 介导的转录程序 [25]。
然后,通过皮下注射 5637 细胞探讨 THZ1 在无胸腺裸鼠体内的抗肿瘤作用。让肿瘤生长 2 周后,将小鼠随机分配到两组(对照组和 THZ1治疗组),发现与对照组相比,THZ1 治疗组的肿瘤大小显著减小。同时,在施用 THZ1后,没有注意到体重显著减轻或其他不良反应。异种移植样品的 H&E 和免疫组织化学(IHC)分析显示 THZ1 强烈抑制细胞增殖并诱导细胞凋亡。这些发现表明 THZ1 在体内对 BC 细胞发挥有效的抗肿瘤作用。
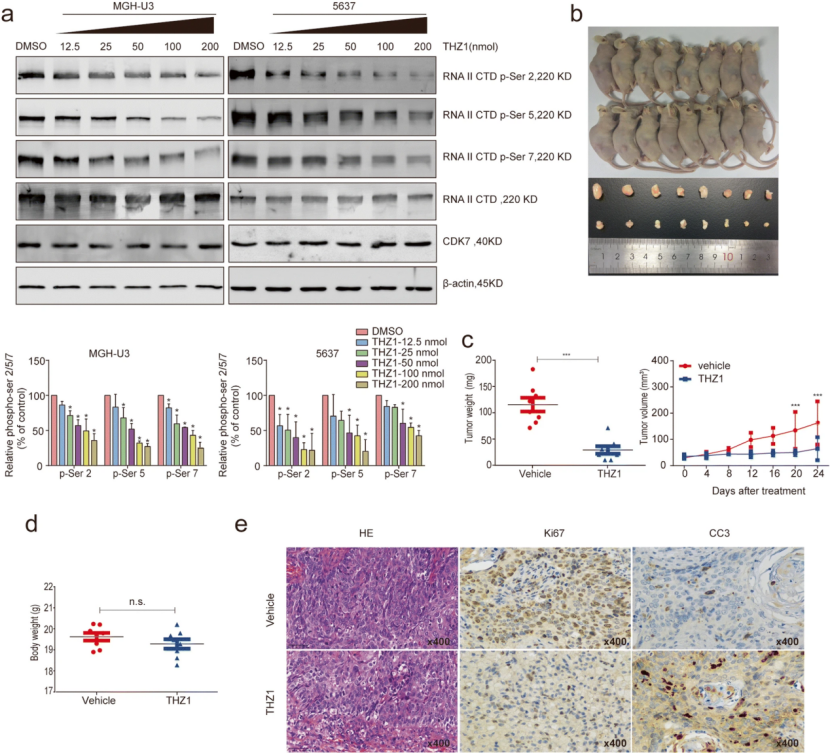
作者接下来探讨THZ1 诱导的 BC 细胞基因表达谱中的转录改变。通过全转录组测序 (RNA-seq) 分析确定了在 MGH-U3 和 5637 细胞中敏感基因的子集(定义为下调的差异表达基因)。在用 50 和 200 nM/L THZ1 处理 6 小时的两个 BC 细胞系的聚集热图中发现,与低剂量 THZ1 相比,高剂量 THZ1 导致全局基因下调。此外,还发现高剂量THZ1和低剂量THZ1处理后,两种不同剂量方案处理的MGH-U3和5637细胞的基因表达变化没有强相关性,表明在两种细胞系中对 50 nM 和 200 nM THZ1 处理的响应存在差异。事实上,之前的一项研究表明 THZ1 可以共价结合并不可逆地抑制 CDK7,但在使用更高浓度的治疗后,可能会对 CDK12 激酶活性产生额外的交叉反应。因此,为了避免这种可能影响 BC 细胞对 CDK7 抑制的转录和表型反应的共同药理学,作者接下来关注低剂量 THZ1 治疗后的变化。作者发现 2227 个抑制基因对低剂量 THZ1 治疗敏感,包括一些众所周知的致癌基因,如 BRCA1、EGFR、TP63、XBP1、CDR2 L 和 GRWD1。对50 nM THZ1 处理的 BC 细胞中下调的前 20% 基因进行了GO 富集分析发现 ,THZ1 敏感基因的子集主要富集在转录调控、DNA 修复方面。
已知超增强子(SE)相关癌基因通常表现出对转录抑制的高度敏感性,在癌症发展中发挥关键作用。为了研究 SE 是否参与促进转录调节因子的过度激活,作者首先使用H3K27ac抗体进行 ChIP-seq 来表征 MGH-U3 细胞中的 SE 景观,同时通过分析公开可用的数据集 (GSE75286) 来表征5637 细胞中的 SE 景观,发现 H3K27ac 信号在两种 BC 细胞系之间显示出高度一致的特征,分别在 MGH-U3 和 5637 细胞系中鉴定了 648 个和 890 个 SE 相关基因。与 TE 亚组相比,SE 组在峰高和密度方面表现出 H3K27ac 信号的特异性和显著富集,发现 SE 相关基因的调节作用在 (i) 肿瘤相关功能中显著丰富,包括细胞周期进程、细胞凋亡和增殖,(ii) 染色质表观遗传景观的磷酸化和异常变化 (iii) 由 RNA 聚合酶 II 介导的转录。通过基因集富集分析(GSEA)发现 ,BC 中 SE 相关基因在两种细胞系中均显著富集 THZ1 敏感转录物。基于这些结果,得出结论,BC 细胞中与 SE 相关的中枢基因过度活跃并且对转录抑制高度敏感。
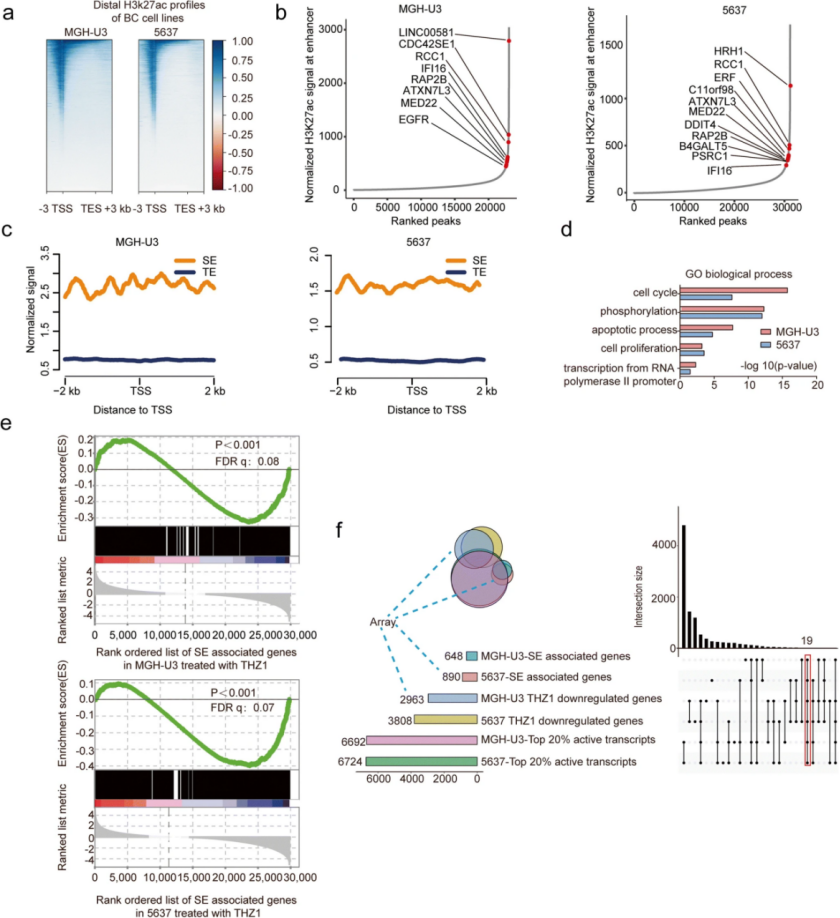
接着,作者对从 BC 细胞获得的 RNA-seq 谱和 SE 谱进行综合分析,经过严格筛选,鉴定出 19 个候选癌基因参与 BC 发病机制。总结了候选致癌基因的 RNA-seq 信号。随后的 qPCR 结果显示,所有 19 个关键癌基因都被证实对 CDK7 抑制过敏(用 50 nM 抑制剂处理 6 小时)。与 CDK7 趋势相似,作者发现 19 个候选基因中有 12 个的高表达与 BC 患者的不良生存显著相关。在 MGH-U3 细胞中通过 CRISPR/Cas9 介导的基因编辑更仔细地分析了选定的基因,发现 8/12 候选基因的沉默显著抑制了 BC 细胞集落形成。由于 MGH-U3 细胞集落形成能力和活力的显著降低,四个基因EGFR、DDIT4、MED22 和 B4GALT5被确定为前 4 个最有希望的候选癌基因。
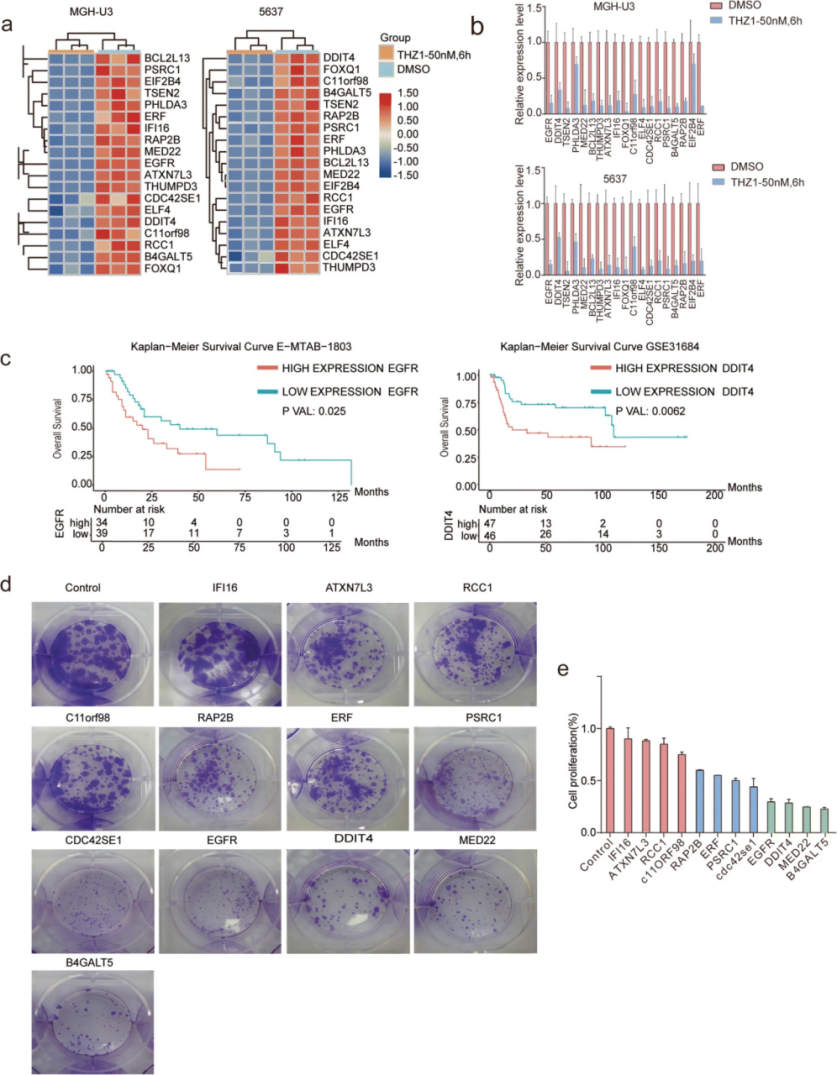
根据 3 年和 5 年总体生存结果的分析发现,4 种选定癌基因的高表达与生存时间缩短显著相关。H3K27ac ChIP-seq 谱显示 4 个候选癌基因与 MGH-U3 和 5637 细胞中 SE 相关基因位点存在结合。通过多重免疫组织化学染色发现,与副肿瘤组织相比,除了 MED22所有蛋白质在 BC 组织中都以相对较高的水平同时表达。
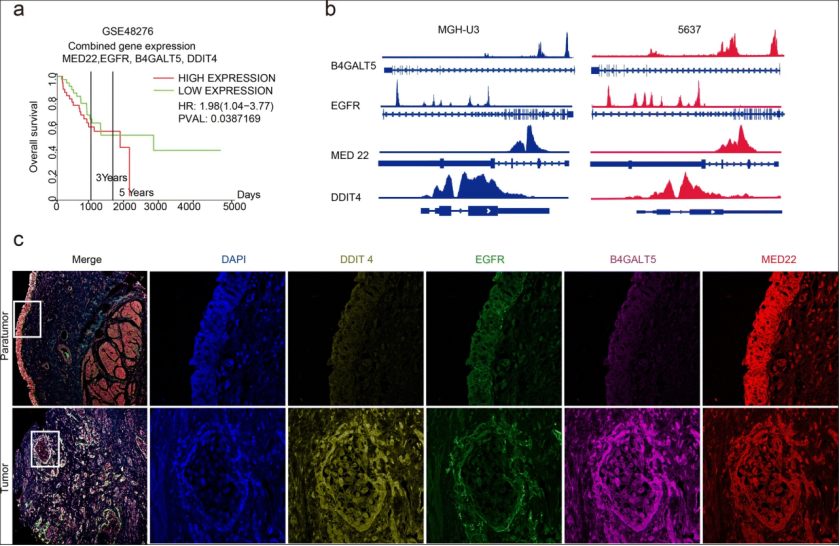
在进一步评估雷帕霉素的联合抗肿瘤作用之前,作者重点研究了 DDIT4 的表达水平与临床病理特征之间的关系,包括性别、年龄、淋巴结转移、存活率和肿瘤分期。在 63 名 BC 患者中发现 DDIT4 表达升高与晚期肿瘤分级之间存在正相关。
作者评估了 THZ1 和雷帕霉素在抑制两种代表性细胞系(MGH-U3 和 5637)增殖和集落形成方面的协同作用。与 THZ1 或雷帕霉素单一治疗组相比,观察到响应于联合治疗的长期增殖和集落形成的抑制增加。同时还发现 5637 细胞比 MGH-U3 细胞对协同处理更敏感。
以顺铂(cis-dichlorodiammine Platinum,cis-DDP)为代表的铂类化疗已成为BC最重要的治疗选择之一,因此作者评估了 cis-DDP 对 MGH-U3 和 5637 细胞的影响,发现它强烈抑制了两种细胞系的生长。接下来,作者将 THZ1 或雷帕霉素与cis- DDP 组合以确定它们的抗生长功效并观察到效果的改善,表明组合治疗的优势。
最后,作者得出结论,THZ1 或雷帕霉素可以增强 BC 细胞对常规化疗的敏感性。
2022年度国自然医学部国自然40大科研热点的中标数统计如下:
2022热点 | 2022中标数 | 2022热点 | 2022中标数 |
免疫调控 | 907 | 中性粒细胞 | 112 |
巨噬细胞 | 591 | 反馈回路 | 104 |
线粒体 | 491 | 乳酸化 | 104 |
血管功能 | 487 | 可变剪接 | 71 |
外泌体 | 470 | AI机器学习 | 67 |
自噬 | 404 | 类器官 | 67 |
铁死亡 | 337 | 炎症小体 | 62 |
干细胞 | 329 | 染色质重塑 | 58 |
代谢重编程 | 325 | 单细胞测序 | 54 |
m6A/m5C/m7G | 320 | 糖基化 | 50 |
泛素化 | 225 | 低氧缺氧 | 50 |
circRNA | 221 | 相分离 | 50 |
lncRNA | 204 | 泛凋亡PANoptosis | 42 |
细胞焦亡 | 175 | 细胞衰老 | 37 |
组蛋白 | 171 | 胞葬 | 33 |
肠道菌群 | 133 | CRISPR | 33 |
乙酰化 | 125 | 增强子 | 29 |
内质网 | 125 | 精氨酸甲基化 | 25 |
转录调控 | 112 | 迁移体 | 8 |
糖酵解 | 112 | 血管拟态 | 8 |
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)