ADC药物的研发并非一帆风顺,从“魔法子弹”概念的提出到现在将近100年的历史,但最终守得云开见月明。DS8201重新定义了ADC,但它不是ADC药物的的终点,而是全新的起点。全面认识ADC及相关技术,可以站在巨人肩膀上看得更远,不限于自己所研究的领域,更可以从全局了解ADC竞争格局。从而实现ADC药物产业的新发展、新突破。
——VIP说、洞见conjugates

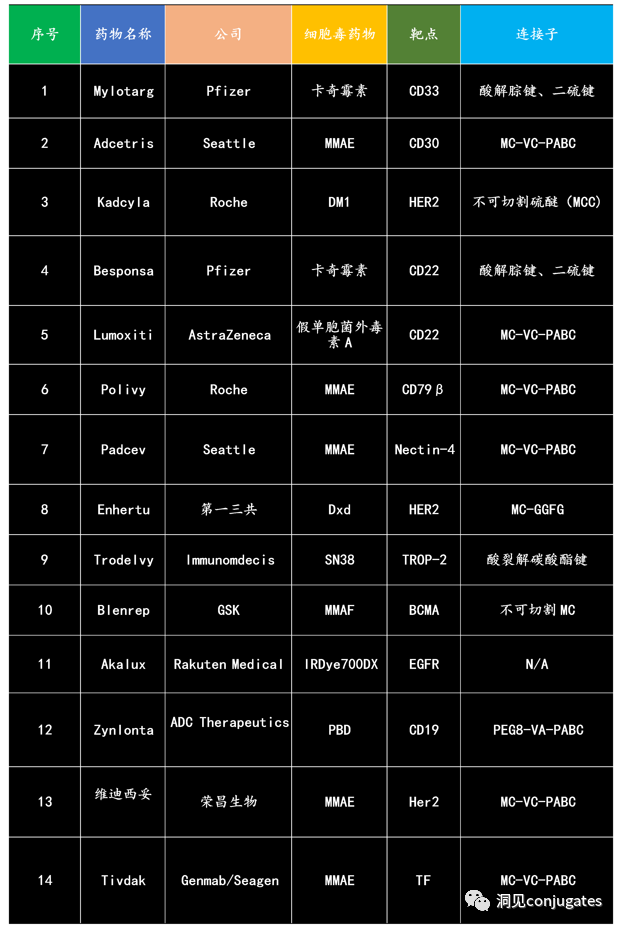
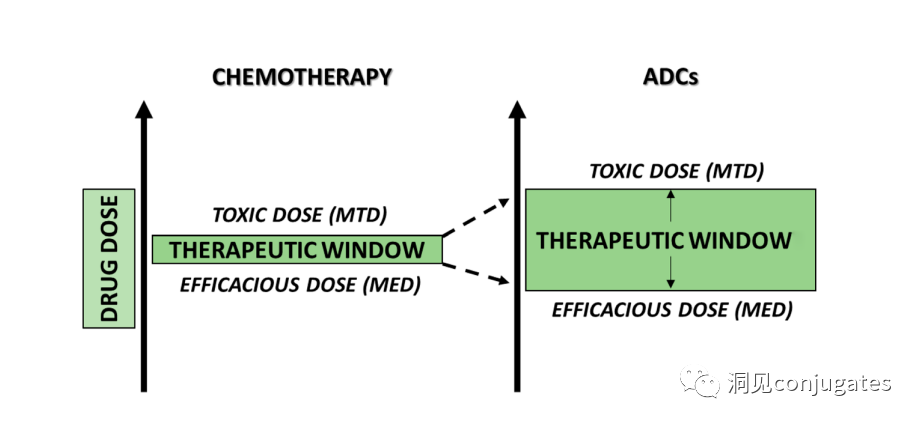
抗体-药物偶联物 (ADC)由linker、payload、单克隆抗体 (mAb) 组成。它结合了高特异性靶向能力和强效杀伤作用的优势,实现了对癌细胞的精准高效杀灭,已成为抗癌药物研发的热点之一。自第一个 ADC 药物Mylotarg ®于2000年获得FDA批准以来,截至2021 年 12 月全球共有 14 款 ADC 药物获批用于血液系统恶性肿瘤和实体瘤,此外,目前还有100多个ADC候选者处于临床试验的不同阶段。

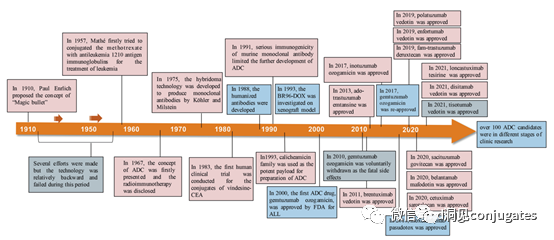
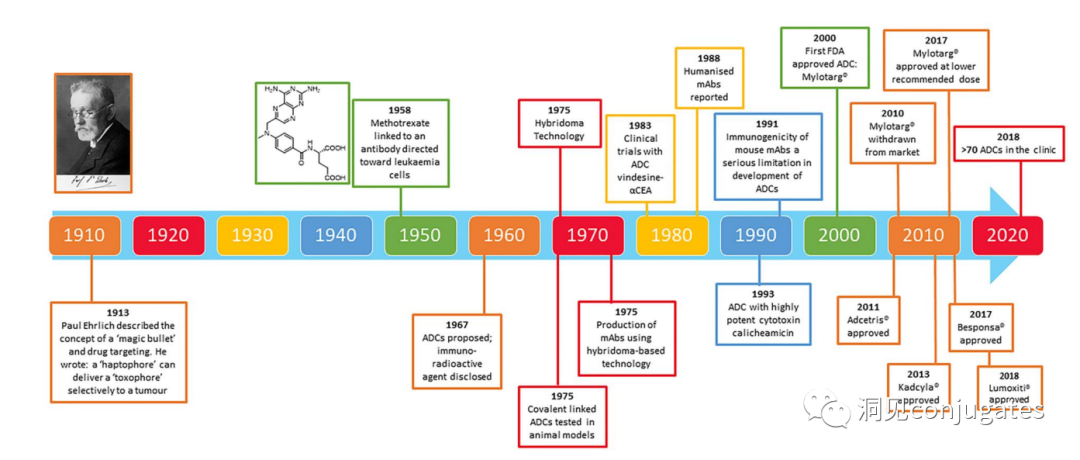
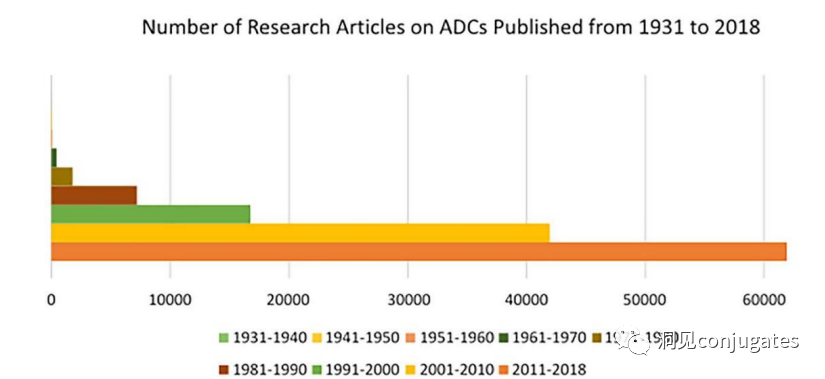
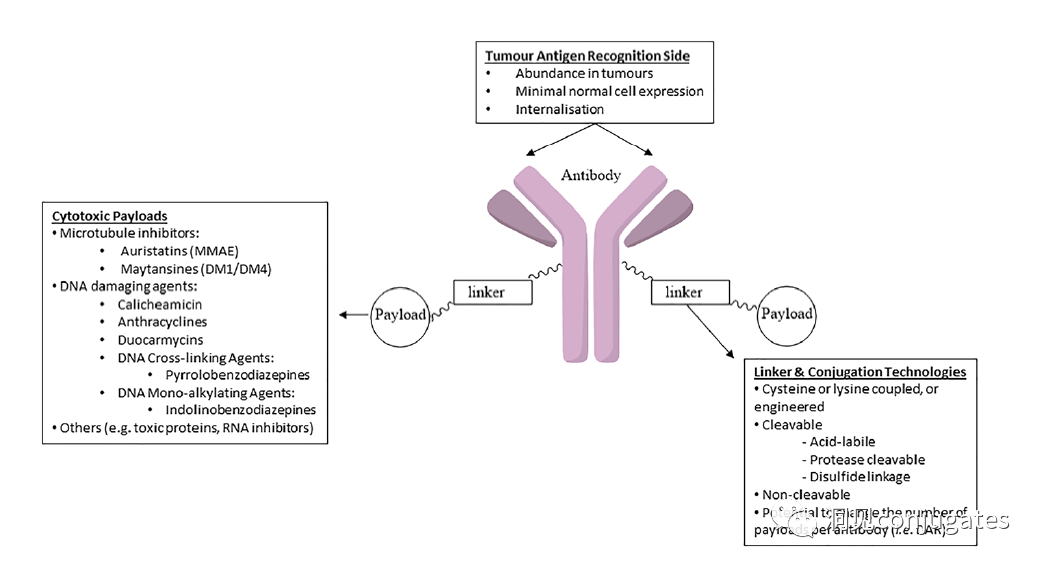
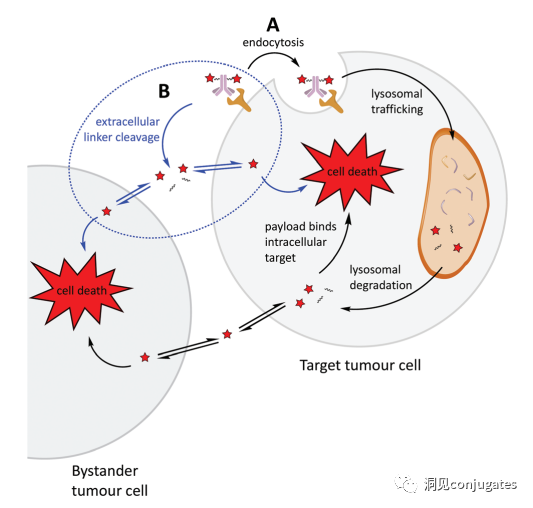
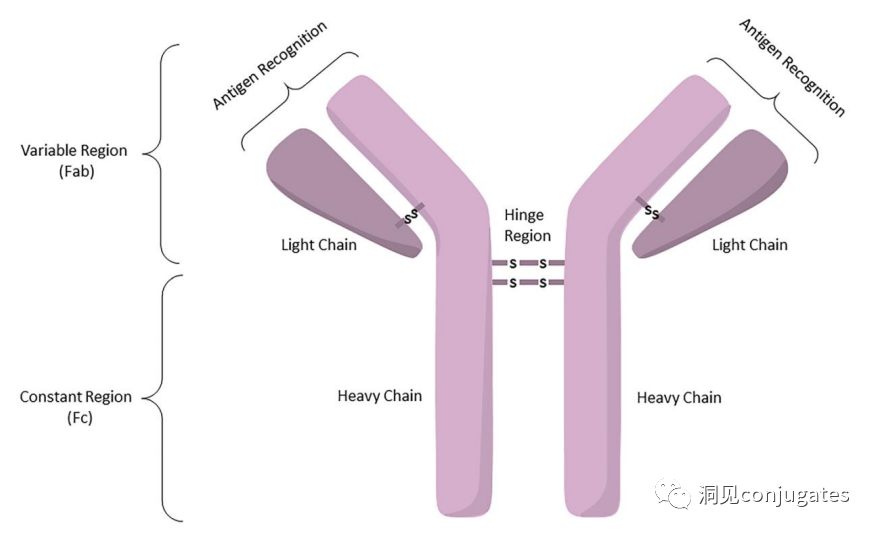
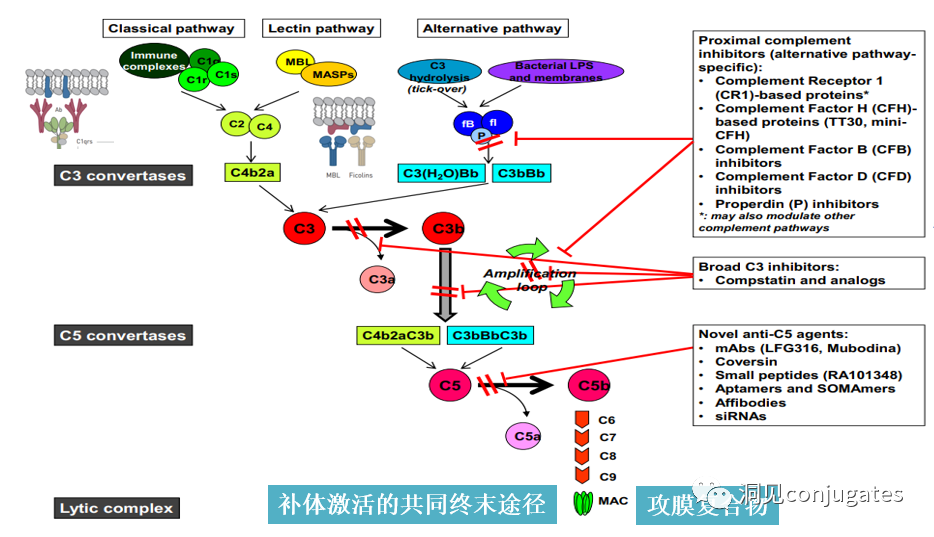
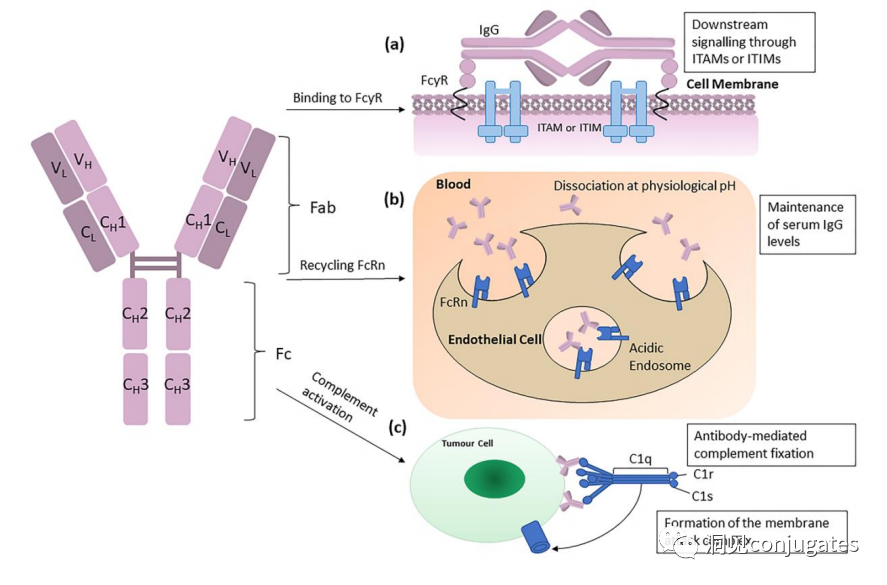
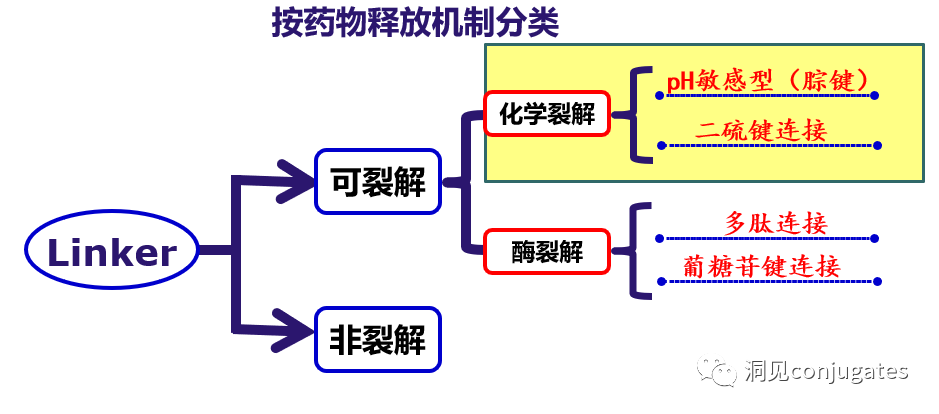
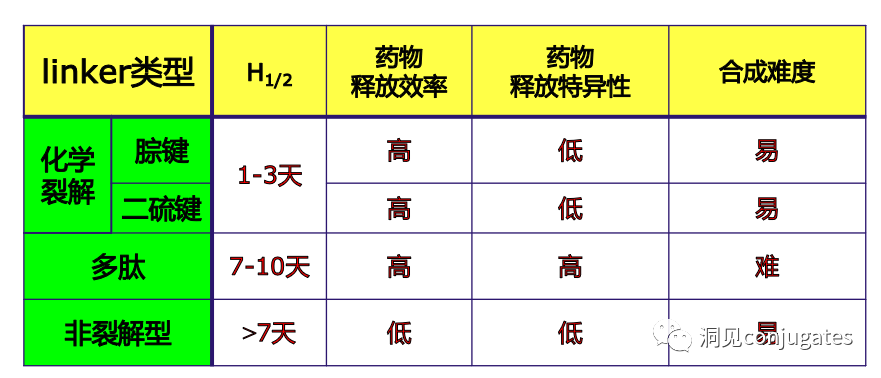
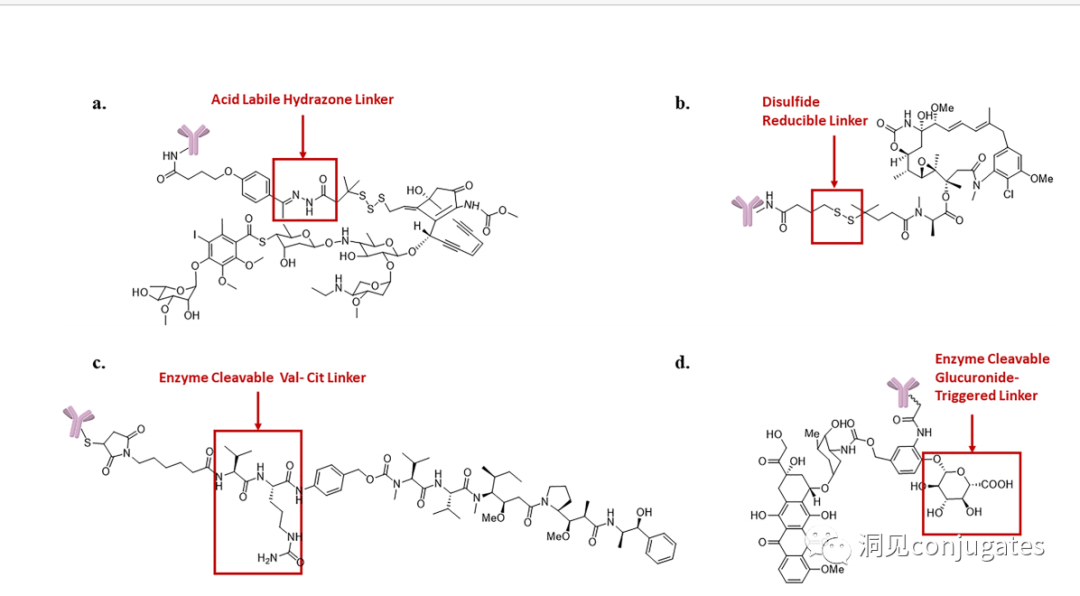
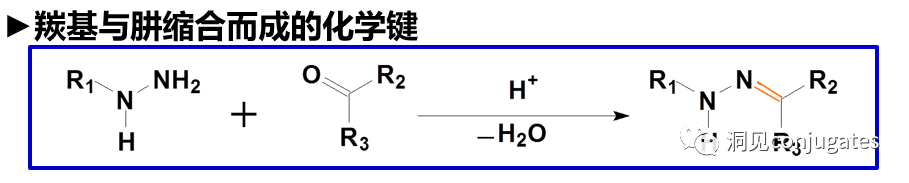
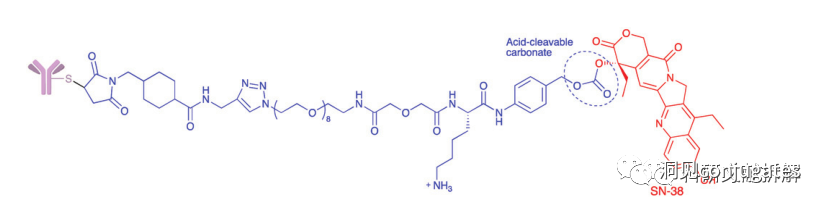
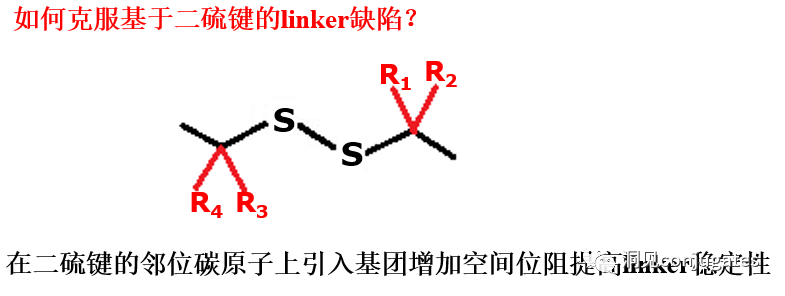
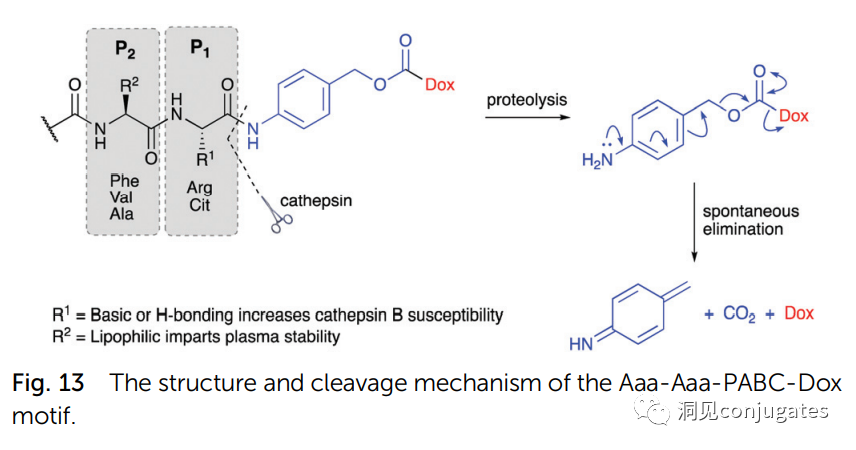
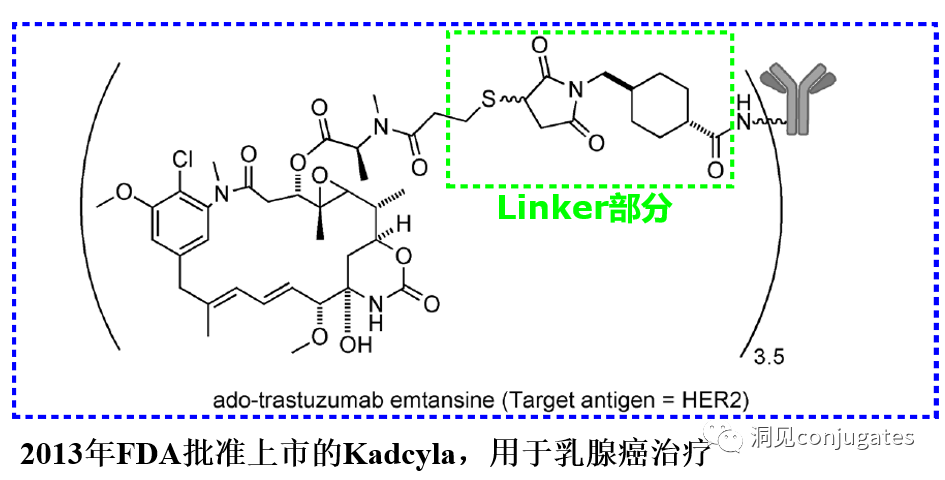
早在 20 世纪初,Paul Ehrlich 就首先提出了“魔法子弹”的概念,并假设某些化合物可以通过某些靶点直接进入肿瘤细胞,从而治愈疾病。从理论上讲,这些化合物应该能有效杀死癌细胞,但对正常细胞无害。2000年,美国食品药品监督管理局(FDA)首次批准ADC药物Mylotarg® (gemtuzumab ozogamicin)用于成人急性髓系白血病(AML),标志着ADC靶向治疗癌症时代的开始。图1描绘了过去百年ADC药物从婴儿阶段到成熟发展阶段的标志性事件。随着靶点和适应症的不断扩大,ADC正在引领癌症靶向治疗的新时代,未来有望替代传统化疗药物。ADC 的概念最初是由 Paul Ehrlich 在 1900 年代初期描述的,他将其描述为“魔法⼦弹”。它们的发展并⾮⼀帆⻛顺,直到 1950 年代初才取得重⼤进展。在 1980 年代和 1990 年代初期 ADC 也⾯临许多挑战。靶抗原选择不佳是早期 ADC 失败的最可能原因,例如⽤于⾮⼩细胞肺癌的 KS1/4 抗体-甲氨蝶呤偶联物和⽤于转移性乳腺癌的 BR96 抗体-多柔⽐星偶联物。这两种ADC均进入了临床,但⼏乎没有提供治疗益处。可能限制这些早期 ADC 成功的其他因素是它们使⽤引发免疫原性反应的嵌合或⿏类抗体,以及使⽤具有低细胞毒性效力的Payload。ADC 研究领域在过去⼏年中发展迅速,⾃ 1970 年以来发表的研究论⽂ 数量说明了这⼀点。很明显,研究⽂章的数量在 2000 年⾄ 2010 年间翻了⼀番,部分原因是 Kadcyla® 和 Adcetris® 在此期间都处于临床开发阶段。2011年⾄2018年间,发表了超过 60000 篇研究⽂章。ADC是通过linker,payload与 mAb相连。ADC与癌细胞表⾯的抗原结合,然后内化,在溶酶体中释放⾼度细胞毒性的Payload分⼦,通常通过溶酶体切割。为了设计成功的 ADC,了解潜在的作⽤机制⾄关重要。⼀个有效的 ADC 需要保留单克隆抗体的选择性,同时能够释放附⾜够⾼浓度Payload以杀死⽬标肿瘤细胞。这些步骤中的每⼀个都涉及多个独特的挑战,使 ADC 的设计变得复杂。ADC 旨在靶向并杀死癌细胞,因此抗体必须能够识别并结合位于肿瘤细胞上的相应抗原。⼀旦与抗原结合,整个抗原-ADC 复合物就会通过受体介导的内吞作⽤被内化。内化过程随着包含 ADC-抗原复合物的⽹格蛋⽩包被的早期内体的形成⽽进行。⼀旦进⼊溶酶体,ADC 就会被降解并释放细胞毒性Payload释放到细胞中,导致细胞死亡。细胞死亡的机制将取决于细胞毒性Payload的类型。例如,微管蛋⽩抑制剂,如 auristatins 或美登素,通过⼲扰微管蛋⽩引起胞质分裂的破坏;⽽ DNA 相互作⽤剂,如 PBD ⼆聚体或卡奇霉素,引起DNA 损伤导致细胞凋亡。⽬前正在开发的新型Payload会⼲扰其他细胞过程,例如 RNA 加⼯。ADC 作⽤机制的⼀个重要⽅⾯是旁观者效应,即游离药物通过细胞膜从肿瘤细胞输出到肿瘤环境。这具有杀死邻近肿瘤细胞的潜在治疗益处,包括那些表⾯上可能没有相关抗原的肿瘤细胞.另⼀个关键是确保⾜够浓度的Payload到达胞内并杀死癌细胞, 实际上,这是⼀个难以保证的复杂过程。据估计,即使 ADC 的整体作⽤机制以 50% 的效率⼯作,也只有 1-2% 的Payload会到达肿瘤细胞。因此,重要的是选择的有效载荷是⾜够的细胞毒性以在⾮常低的浓度下发挥作⽤。现在已经认识到,抗原靶点、抗体、Linker和Payload成分的选择,以及它们如何有效地协同⼯作,对于ADC的成功⾄关重要。链接子在细胞外的肿瘤微环境中断裂释放出载荷,不需要细胞的内吞作用,可以选择非内化抗原作为靶点。设计ADC最关键的因素之⼀是抗体的选择。最重要的是对抗原具有⾼特异性。缺乏⾼特异性并与其他抗原结合的抗体可能会产⽣不可预测的作⽤,例如通过与健康组织相互作⽤导致脱靶毒性或在到达肿瘤部位之前过早消除。抗体以⾼亲和力与靶抗原结合同时低免疫原性也很重要。另⼀个重要特征是有利的药代动力学 (PK) 特性。最后,如果 mAb 通过直接调节靶抗原的⽣物活性⽽具有固有的抗肿瘤活性,这将是有帮助的,例如抗⼈表皮生长因子受体的情况2 (HER2) mAb 曲妥珠单抗 (Herceptin®),它是曲妥珠单抗 emtansine (Kadcyla®) 的抗体成分。抗体根据其重链恒定区的序列分为五类:免疫球蛋⽩ M (IgM)、IgD、IgG、IgE 和 IgA。在这 五类中,IgG 最常⽤于癌症免疫疗法。典型的 IgG1 抗体由两条重链 (H) 和两条轻链 (L) 组成, 其中包括构成 Fc 结构域的恒定 (C) 区和构成提供抗原特异性的 Fab 结构域的可变 (V) 区。Fc区被免疫细胞识别。Fc 片段不识别相应的抗原,⽽是结合各种细胞受体(例如T 细胞)和补体蛋⽩。所有抗体都在其恒定区保守位置具有糖基化位点。例如,在 Fc 区N297 残基上拥有⼀个N-糖基化位点。IgG 的亚类,更具体地说是 IgG1 和 IgG3,是经典补体途径的有效激活剂。两个或多个 IgG 分⼦与细胞表⾯的补体成分 1q (C1q) 结合, 1q (C1q)与Fc 结构域⾼亲和力结合随后激活 C1r 酶活性,随后激活下游补体蛋⽩。这种级联反应的结果是膜攻击复合物 (MAC) 在肿瘤细胞表⾯形成孔隙,随后肿瘤细胞裂解。为了保证有效的内化,抗原结合位点应具有尽可能⾼的亲和力(目前研究发现亲和力并不是越高越好,过高的亲和力存在实体瘤BSB,如果靶点在肿瘤组织中的表达异常,且亲和力高,需要大剂量ADC去饱和肿瘤边缘的可用受体,从而释放ADC 分子可以继续渗透到更深的层,结合位点的屏障效应唯一可以修改的就是结合亲和力,在肿瘤微环境,抗体药物更容易被第一个接触的靶点所捕获,而非渗透到更远的位置。所以,根据肿瘤大小优化亲和力,以确保有足够的亲和力可以进行药物内化,也要让药物可以渗透到肿瘤的更深处,需要一个平衡)。下图显⽰了IgG 抗体的抗原如何与 Fc受体结合并通过基于免疫受体酪氨酸的激活基序 (ITAM)或基于免疫受体酪氨酸的抑制基序 (ITIM)启动信号。IgG 可以与内皮细胞上的Fc 受体 (FcRn) 结合以维持⾎清 IgG ⽔平,并与肿瘤细胞结合,在那里它们可以募集补体成分 1q (C1q) 以启动补体级联反应,从⽽导致肿瘤细胞被 MAC 裂解。考虑到肿瘤部位所需的⾼特异性、亲和力和⻓时间暴露,理想情况下,抗体选择应确保与健康组织的相互作用最⼩、对靶抗原的亚纳摩尔亲和力和较⻓的药代动力学半衰期,具有最⼩的免疫原性。ADC 的免疫原性程度是⼀个关键因素,也是循环半衰期的重要决定因素。尤其是,缺乏肿瘤特异性的抗体可能会由于免疫原性⽽迅速从循环中清除,从⽽导致治疗效果下降。在 ADC 发展的早期,研究是基于⼩⿏的单克隆抗体,导致在单次给药后⼏周内形成⼈抗⼩⿏抗体。因此,⿏抗很快被嵌合 IgG 抗体取代,随后是⼈源化 IgG。近年来,ADC 主要基于全⼈源抗体。使⽤ mAb ⽽不是⼩分⼦化学治疗剂进行癌症治疗的最重要的好处之⼀是基于 mAb 的药物可以在持续时间、代谢和消除⽅⾯具有良好的药代动力学。如上所述优化。⼀旦将 mAb 给药到⾎流中,它们可以通过内⽪中的孔外渗或通过胞饮作 ⽤分布到肿瘤组织中。ADC 在肿瘤组织中的分布受到抗体⼤⼩的限制,抗体⼤⼩通常占到 ADC 质量的95%。然⽽,与具有单层内⽪细胞彼此紧密连接的正常⾎管不同,肿瘤内⽪通常以过度分枝和发芽为特征,从⽽导致“渗漏”单层。因此,ADCs虽然分子量较大但仍然可能通过渗漏的脉管系统分布到肿瘤组织中,但它们在肝脏、肠道、肌⾁和⽪肤等代谢和清除器官中的分布受到限制。最近,正在研究抗体⽚段,由于它们的尺⼨更⼩,肿瘤组织穿透能力更强,与全尺⼨抗体相⽐,它们通常具有更短的半衰期。Linker的设计、结构和化学性质对 ADC 的特性至关重要,这关系到整个ADC药物的特异性、有效性和安全性。通常,linker设计为在⾎流中稳定,在肿瘤部位能有效释放Payload。偶联物在缓冲⽔溶液中保持稳定也是⾄关重要的。连接剂根据其Payload释放机制分为不同类型,主要的两种是“可裂解”或“不可裂解”(推荐阅读:ADC药物中常见linker种类及体内切除机理)。可切割的Linker利⽤⾎流和肿瘤细胞内细胞质之间的条件差异。ADC-抗原复合物内化后的环境变化触发Linker的切割和活性Payload的释放。可切割的Linker分为三个主要⼦类别:(1)酸不稳定(例如腙),(2)可还原(例如⼆硫化物)和(3)酶可裂解(例如肽)。下图显⽰了在 Gemtuzumab ozogamicin (Mylotarg®) 中使⽤的腙接 头的⼀个例⼦。Payload的非特定释放,可能导致全⾝毒性。这也是Mylotarg在2010年⾸次退出市场的原因之⼀另⼀种类型的酶可切割Linker基于 β-葡糖苷酸部分。β-葡萄糖醛酸酶(溶酶体中)可以从含有β-葡萄糖苷酸的Linker释放Payload,并且在某些肿瘤细胞类型中过度表达。β-葡萄糖苷酸Linker的⼀个重要特征是其亲⽔性,与含有基于⼆肽或其他接头类型的结构相⽐,它可以潜在地减少偶联过程中的聚集。β-葡萄糖苷酸已被⽤作Linker部分来连接Payload,包括 auristatin 衍⽣物MMAE和 MMAF以及阿霉素。中性的血液环境中(pH7.3-7.5):约6%的裂解率 胞内溶酶体(pH4.5-5.0)或者内涵体(pH5.0-6.5),易裂解,在pH4.5时裂解率约为97%。另一种常用的酸条件水解的结构还有碳酸酯类linker。简单的碳酸酯在血清中的稳定性有限,需要加入一个对氨基苄基(PAB)可以显著提高半衰期。碳酸酯结构在酸性条件下水解释放SN-38药物和二氧化碳。二硫化合物在生理PH条件下稳定,但是容易受到硫醇的亲核进攻,从而切断链接(包含位阻二硫键)。血液中含巯基化合物比较复杂,还有白蛋白的干扰,含有自由巯基的小分子物质(GSH和Cys):细胞内的溶酶体和细胞外的肿瘤微环境中均存在特异性的酶可以选择性裂解相应的底物。基于多肽的Linker:利用溶酶体中的蛋白质水解酶如Cathepsin 和plasmin(这些蛋白质水解酶活性在血液环境中被严格抑制,而在溶酶体环境中酶活性最佳)►基于多肽的Linker血液稳定性更高,在癌细胞中裂解更快最佳的二肽:Val-Cit和Phe-Lys(瓜氨酸-缬氨酸and苯丙氨酸-赖氨酸), 可被Cathepsin B特异酶切水解二肽类的linker通常以组织蛋白酶B作为靶标,组织蛋白酶主要存在溶酶体中,在细胞外也观察到活性。β-葡萄糖醛酸酶是糖苷酶类中的水解酶,利用溶酶体中的β-葡糖苷酸酶催化多糖中β-葡萄糖醛酸残基的分解。►β-葡糖苷酸酶大量存在于溶酶体中,且在某些癌细胞中过表达.►溶酶体环境β-葡糖苷酸酶活性最高,而在血液环境中,酶活受到抑制.葡萄糖醛酸连接子还有一个优势,由于其很高的亲水性,可以大大减少ADC的聚集,这样可以合成高负载载荷的ADC药物(DAR=8)。β-半乳糖苷酶可裂解Linker和葡萄糖醛酸酶类似。
溶酶体中存在酸性焦磷酸酶和酸性磷酸酶,可以水解磷酸酯键。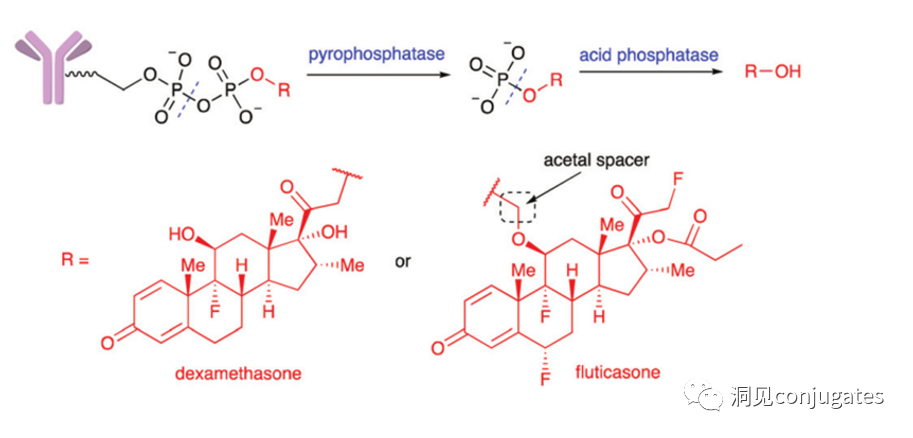 Merck设计了一种二磷酸酯linker,在溶酶体中水解释放原型糖皮质激素类载荷,当测试单磷酸酯链接子时,药物释放相对减缓。ADC被吞噬到细胞后,进入溶酶体途径,在各种裂解酶的作用下,待单抗降解完全后,释放药物的衍生物,对靶细胞进行杀伤。一般稳定性比较好(2)ADC胞内降解后的药物衍生物也应具有可观的细胞毒性,所以该linker的使用对药物的要求比较苛刻。(1)对药物要求苛刻,需要筛选验证药物衍生物的细胞毒性(3)一般ADC胞内降解后的药物衍生物水溶性较好,甚至带有荷电基团,不利于透过细胞膜发挥旁观者效应(Bystander Kill Effect)。
Merck设计了一种二磷酸酯linker,在溶酶体中水解释放原型糖皮质激素类载荷,当测试单磷酸酯链接子时,药物释放相对减缓。ADC被吞噬到细胞后,进入溶酶体途径,在各种裂解酶的作用下,待单抗降解完全后,释放药物的衍生物,对靶细胞进行杀伤。一般稳定性比较好(2)ADC胞内降解后的药物衍生物也应具有可观的细胞毒性,所以该linker的使用对药物的要求比较苛刻。(1)对药物要求苛刻,需要筛选验证药物衍生物的细胞毒性(3)一般ADC胞内降解后的药物衍生物水溶性较好,甚至带有荷电基团,不利于透过细胞膜发挥旁观者效应(Bystander Kill Effect)。
这一期讲到了ADC的全局概况、ADC作用机制、ADC药物设计的复杂性、抗体的选择、linker的选择,那么payload如何选择,偶联方式如何,我们在下一期介绍。
参考文献:
1.Cytotoxic payloads for antibody-drug conjugates
2.其它公开资料整理
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。











































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)