
2022-11-24 来源 : 学术查
国自然热点:免疫炎症
研究背景
破骨细胞(osteolast)引起的炎症性骨丢失是许多炎症性疾病如类风湿性关节炎(RA)、牙周炎和银屑病关节炎等的标志病症。现已证实核因子受体激活剂B配体(RANKL)是生理性破骨细胞分化的诱导剂,但病理条件下炎症性破骨细胞分化的具体驱动因素和机制仍不清楚。因此,有必要对介导这些炎症以及破骨细胞生成的机制展开研究,为类似病症的治疗提供新的思路,以抑制炎症性骨丢失,同时消除或尽量减少对骨重建或生理疾病环境中的免疫反应。
最近几年免疫炎症研究有关的高分文章大量见刊,且国自然项目中与免疫炎症研究相关的项目数量不断增加。免疫炎症在生命科学/基础医学研究热点中的地位可见一斑。
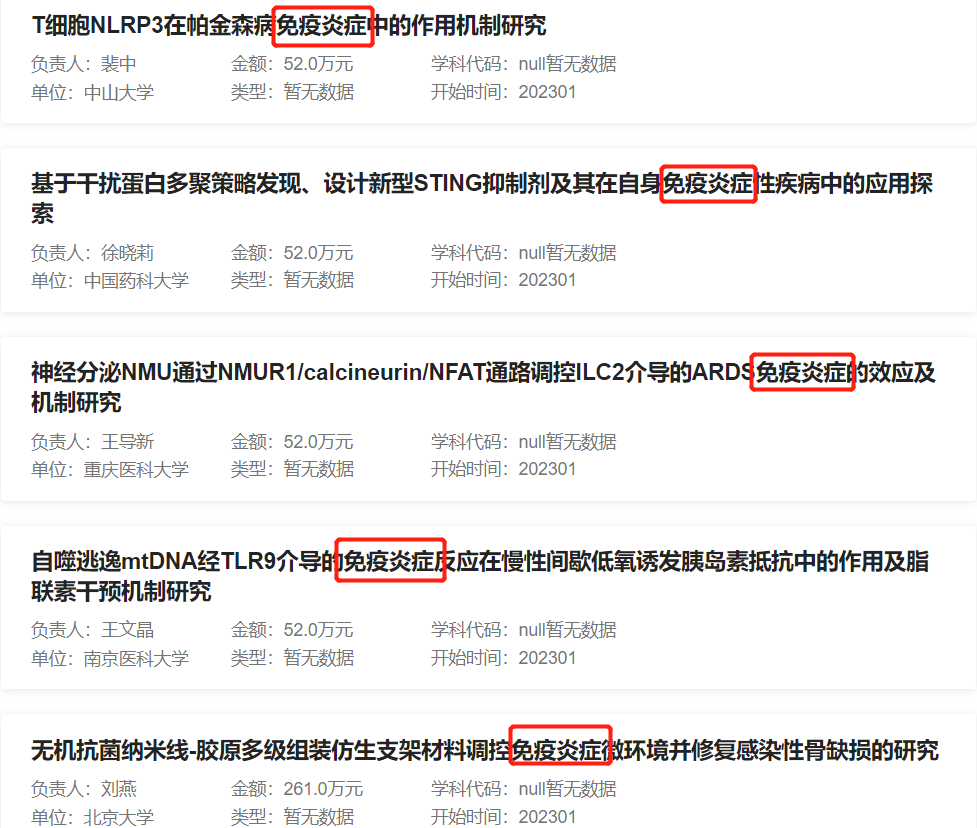
2022年免疫炎症相关部分资助项目列表
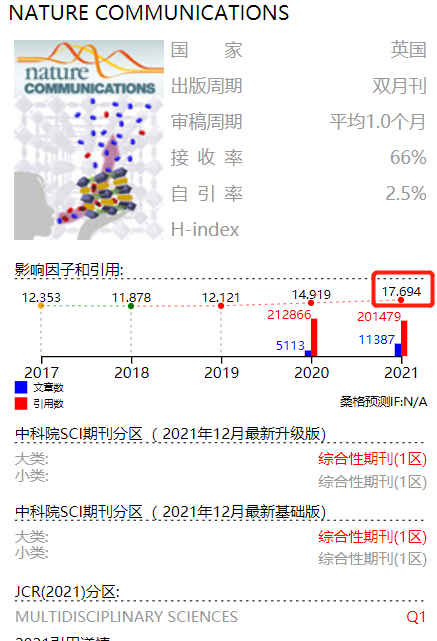
来自关节炎和组织退化项目/美国纽约特殊外科医院David Z.Rosensweig基因组学研究中心的研究团队于2022年7月7日在Nature communications(IF 17.69) 杂志上发表了题为“TGFβ reprograms TNF stimulation of macrophages towards a non-canonical pathway driving inflammatory osteoclastogenesis”的研究论文。主要揭示了TGFβ通过抑制细胞因子IL-1B和IL-6在TNF刺激的巨噬细胞中的表达,抑制TNF的炎症作用,TGFβ的启动强烈增加染色质可及性和H3K4me3/H3K27ac信号,同时减少OC基因位点上的抑制性H3K27me3标记,重新编程巨噬细胞对破骨细胞的作用途径。为治疗炎症性骨吸收提供了新的思路。
原文链接:
https://www.nature.com/articles/s41467-022-31475-1#citeas
主要研究结果如下:
1. TGFβ通过重新编程TNF对巨噬细胞的作用,驱动破骨细胞生成
仅靠TNF(肿瘤坏死因子)未能诱导破骨细胞分化(图1a)。值得注意的是,TGFβ对TNF启动效应与供体之间高度一致(图1b)。另一方面, TGFβ启动几乎完全消除了巨噬菌体的吞噬特性(图1f)。TGFβ启动也强烈抑制了细胞因子IL-1B和IL-6在TNF刺激后的巨噬细胞中的表达(图1g)。这些结果共同表明TGFβ启动抑制TNF的炎症作用,并重新编程巨噬细胞对破骨细胞生成的反应(图1 h)。
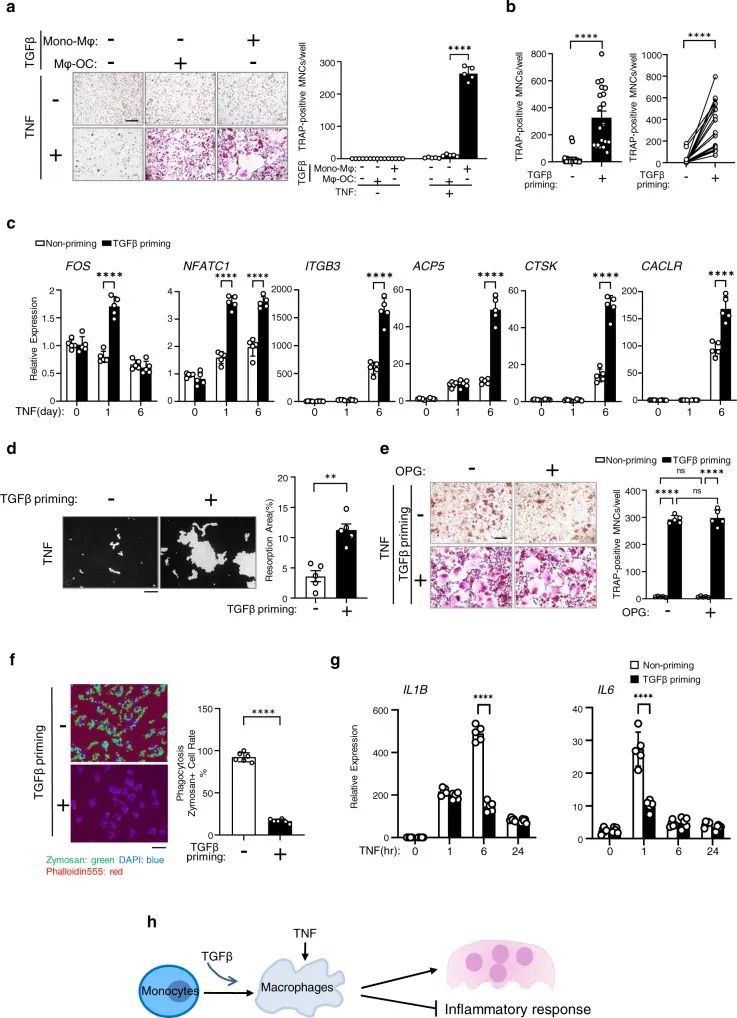
图 1 TGFβ启动开关TNF的作用,驱动破骨细胞生成
2. TGFβ信号的缺乏可阻止TNF,但不能阻止RANKL诱导的破骨细胞生成和骨吸收
作者首先使用骨髓来源的巨噬细胞(BMMs)检测RANKL诱导的破骨细胞生成,发现Tgfbr2ΔM (髓系巨噬细胞和破骨细胞前体中TGFβII型受体被敲除的小鼠)BMMs中的TGFβ信号缺失没有影响RANKL诱导的破骨细胞分化(图2a)。为了测试TGFβ对体内TNF反应的影响,作者采用了一种成熟的小鼠颅骨骨溶解模型进行TNF的炎症反应,PBS注射为阴性对照,在颅骨表面没有观察到吸收坑的形成,髓系中的Tgfbr2缺乏不影响基础破骨细胞生成(图2f,g,h)。结果表明内源性TGFβ信号传导不影响RANKL诱导的破骨细胞形成,但在促进TNF介导的骨形成和加剧炎症性骨吸收方面起着至关重要的作用。
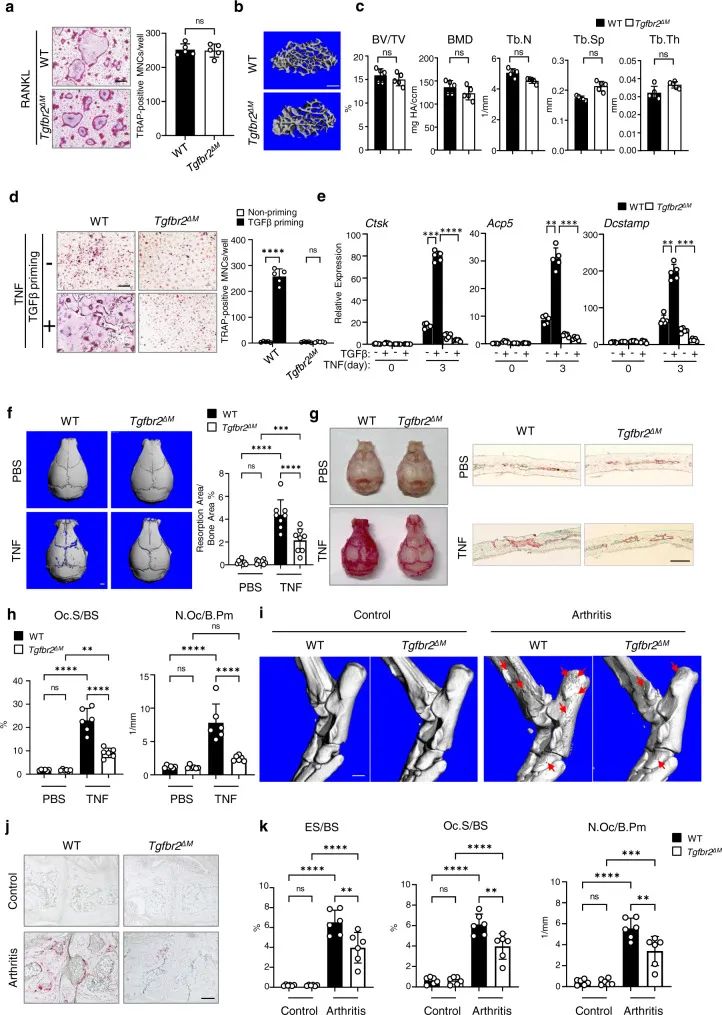
图 2 TGFβ信号转导的缺失抑制炎症破骨细胞生成和骨吸收
★
3.TGFβ的启动抑制了IFN刺激基因的表达,同时转换TNF以诱导破骨细胞基因生成
基于RNA序列的表达热图显示许多干扰素刺激基因(ISG),包括I型干扰素反应基因(图3c)和趋化因子基因(图3d)在刺激24小时后仅受TNF诱导。在TGFβ启动条件下,TNF并未诱导细胞凋亡这些炎症基因反而激活了许多典型破骨细胞基因(以下简称OC基因),以及一组晚期破骨细胞标记物基因(图3e),基因集富集分析(GSEA)也证实I型IFN应答基因和趋化因子基因包括肿瘤坏死因子刺激的顶端巨噬细胞基因集富集显著,而TGFβ诱导的TNF则显示破骨细胞基因集富集(图3f-h)。
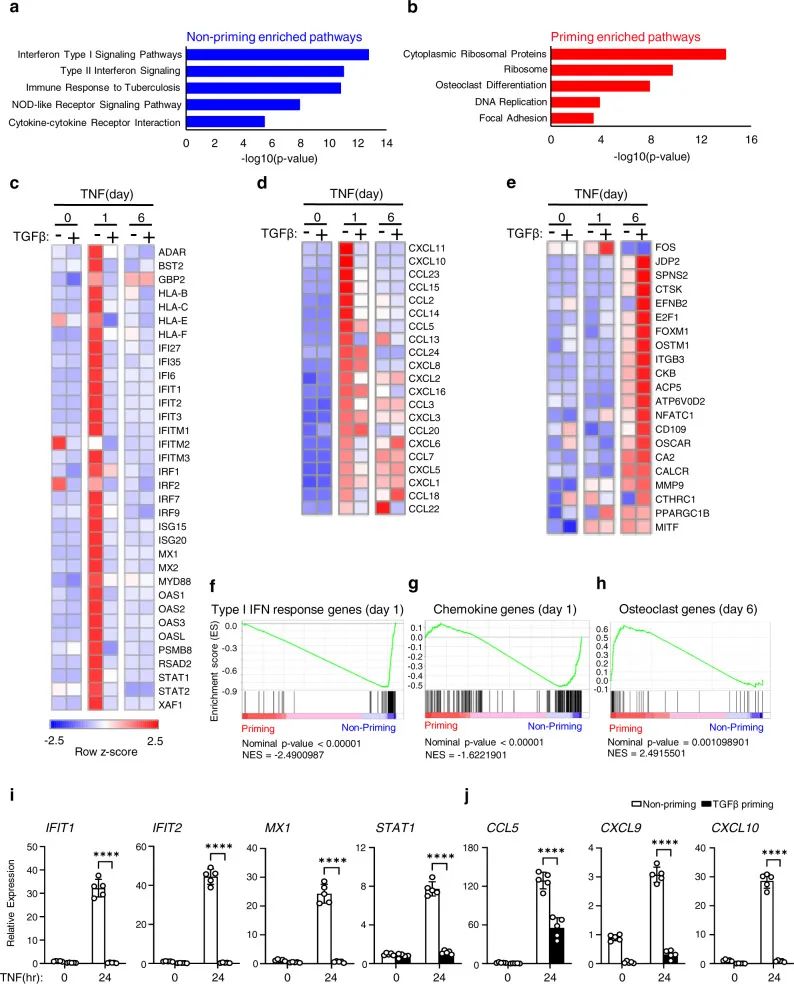
图 3 TGFβ启动重编程TNF对巨噬细胞
转录组的作用
4. TGFβ通过重塑染色质可及性和组蛋白标记,来重编程破骨细胞的基因转录
通过ATAC-seq,发现TNF显著诱导TGFβ非启动和启动条件(图4a)。基因通路分析之后对ISG和OC位点进行评估,在所有ISG中观察到TGFβ诱导的染色质闭合位点。值得注意的是,TNF本身不会改变OC基因位点的染色质可及性(图4e,右图)表明仅靠TNF并不能有效诱导OC基因表达(图3e)。另一方面,TGFβ启动强烈增加染色质可及性和H3K4me3/H3K27ac信号,同时减少OC基因位点上的抑制性H3K27me3标记。这种强大的TGFβ启动为诱导染色质重塑不同的基因提供了必要的先决条件和基因表达程序重新编程的基础,导致TNF有效诱导OC基因的转录发生在巨噬细胞而不是ISGs(干扰素刺激的基因)中。
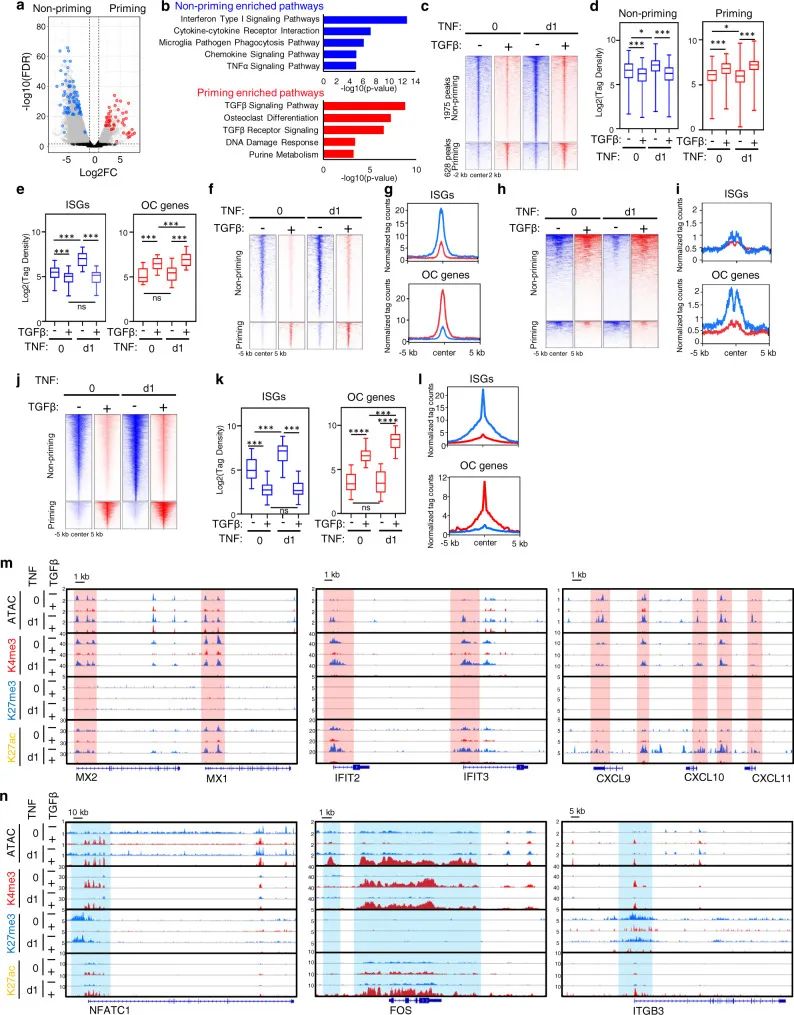
图 4 TGFβ启动调节染色质可及性和组蛋白修饰
5. TGFβ的启动抑制TNF诱导的IRF1-IFNβ轴
接下来,作者对与调节TGFβ启动的基因表达有关的染色质重塑的转录因子和途径进行研究。发现TNF通过内源性IFNβ生成诱导ISGs,此外,还发现TGFβ启动对IFNβ的影响与ISG位点的作用类似,TGFβ启动染色质可及性和IFNB1启动子3′下游的H3K4me3/H3K27ac调节区域(图5b)。因此,TGFβ启动几乎完全消除了TNF诱导的IFNB1基因表达(图5c)和蛋白质产生(图5d)。
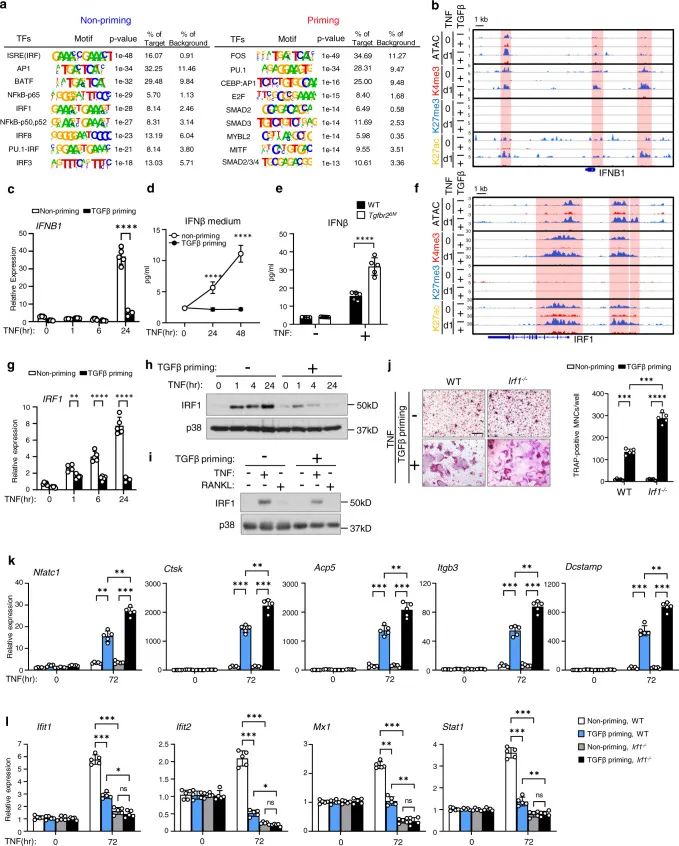
图 5 TGFβ启动抑制TNF诱导的IRF1-IFNβ-ISG轴
6. TGFβ的启动促进IRF8蛋白降解
IRF8是一个破骨细胞分化的有效转录抑制因子,IRF8表达下调是破骨细胞生成的先决条件。TNF单独作用没有显著性差异降低IRF8蛋白水平,但TGFβ启动使TNF能够快速、实质性下调IRF8蛋白(图6a)。有趣的是,TGFβ对大多数其它基因的启动效应作用在转录水平(图6b)但是TGFβ启动并没有通过TNF影响IRF8 mRNA水平的降低。
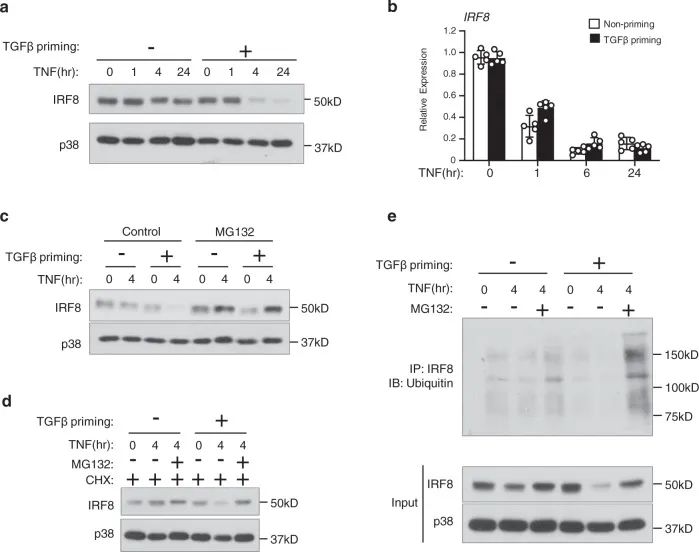
图 6 TGFβ启动促进IRF8泛素化和降解以响应TNF
7. TGFβ的启动使TNF能够诱导不同于RANKL诱导的非典型破骨细胞生成调节因子
作者进一步对TGFβ启动/TNF诱导的ATAC-seq峰下富集的序列进行从头基序分析,发现常见破骨细胞结合位点的富集转录因子E2F1、PRDM1、NFATC1、FOS和MITF(图7c)。不同于这些经典OC转录因子,该研究发现一种异常丰富的MYBL2的转录因子结合位点(编码B-Myb)(图7c)。TGFβ启动显著打开MYBL2启动子和增强子区域的染色质,降低H3K27me3标记,增强H3K4me3/H3K27ac信号(图7d)。MYBL2在基础水平的表达较低,TGFβ启动显著升高TNF诱导的MYBL2表达(图7e,f)。
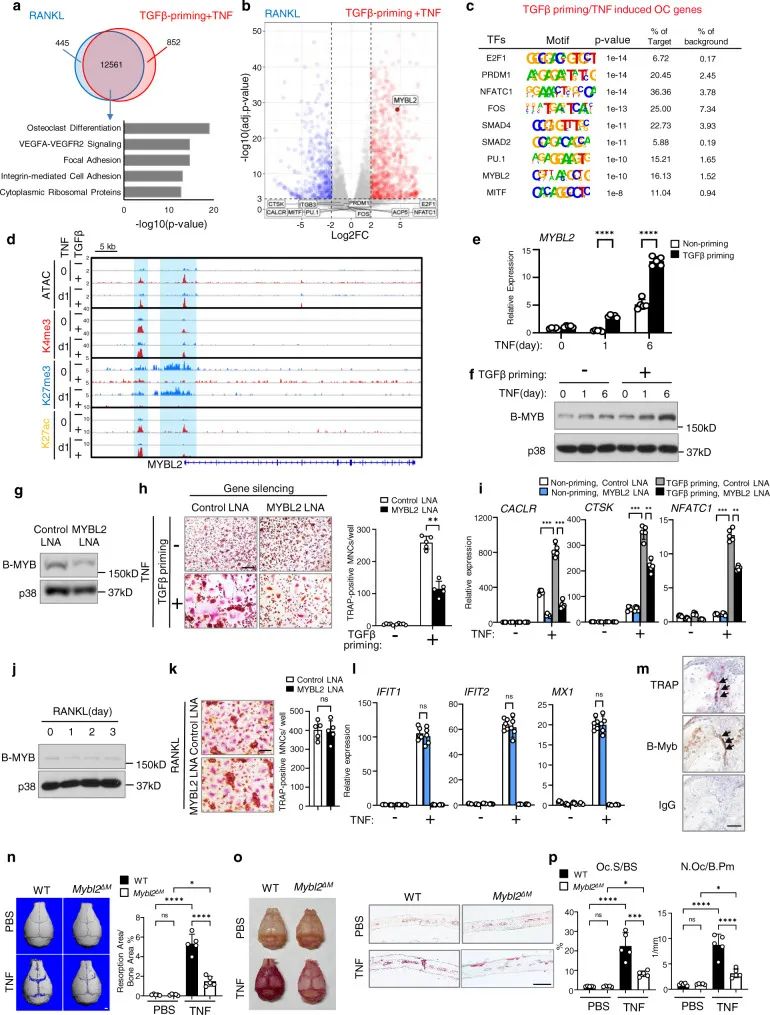
图 7 B-Myb参与TGFβ启动/TNF介导的破骨细胞生成
8. TGFβ表达与RA破骨细胞高度相关
信号通路分析RA和SLE(系统性红斑狼疮)中的差异表达基因(DEGs)显示了不同的相关途径(图8a-c)。与RA最显著相关的途径是TGFβ信号传导(图8c)。基因表达热图(图8d)GSEA分析(图8e)进一步证实了在RA PBMC(外周血单核细胞)中显著富集OC基因。此外,模型系统研究中,RA患者的TGFB1,FOXM1以及MYBL2的mRNA 表达水平显著高于SLE患者;相反,与SLE患者相比,RA患者的IFNB1、IRF1、和IRF8的mRNA表达水平显著降低(图8g)。
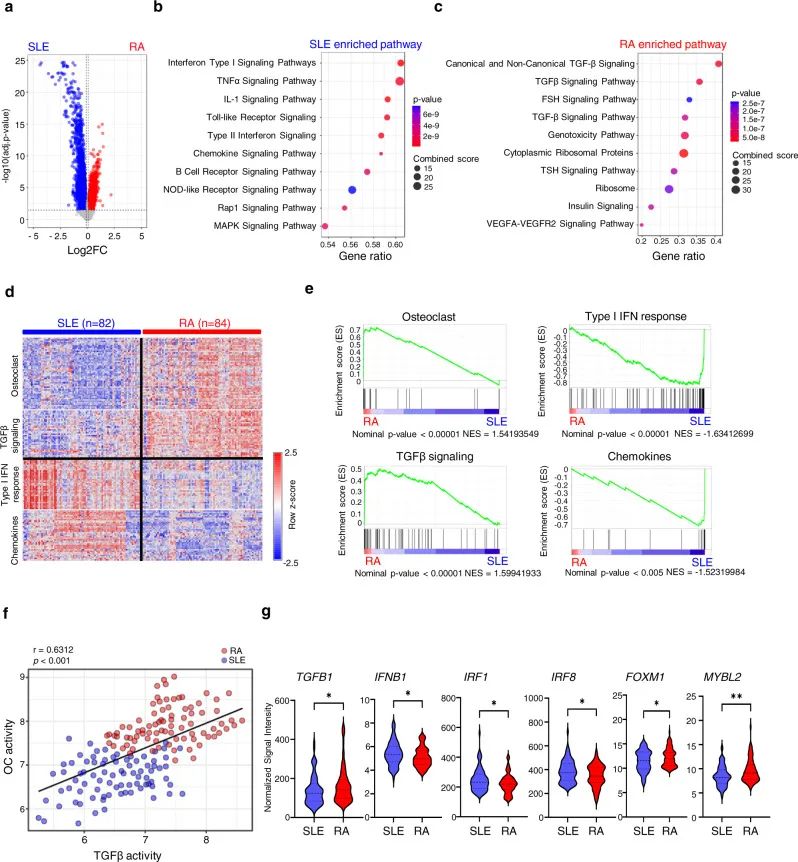
图 8 不同的TGFβ水平/活性导致RA和SLE患者破骨细胞活性不同
小结:
本研究发现TGFβ能够重新编程巨噬细胞对TNF的反应,通过重组染色质和组蛋白修饰改变破骨细胞生成的细胞命运。这些发现明确了之前转化生长因子β在调节肿瘤坏死因子对巨噬细胞极化/分化过程以及RANKL非依赖性破骨细胞分化途径中的未被认识的功能。在TGFβ和TNF介导的炎症反应中发现的这些机制,可以用于炎症性骨吸收治疗,能够选择性地抑制炎症性骨吸收,且不影响生理性骨重塑。
2022年度国自然医学部国自然40大科研热点的中标数统计如下:
2022热点 | 2022中标数 | 2022热点 | 2022中标数 |
免疫调控 | 907 | 中性粒细胞 | 112 |
巨噬细胞 | 591 | 反馈回路 | 104 |
线粒体 | 491 | 乳酸化 | 104 |
血管功能 | 487 | 可变剪接 | 71 |
外泌体 | 470 | AI机器学习 | 67 |
自噬 | 404 | 类器官 | 67 |
铁死亡 | 337 | 炎症小体 | 62 |
干细胞 | 329 | 染色质重塑 | 58 |
代谢重编程 | 325 | 单细胞测序 | 54 |
m6A/m5C/m7G | 320 | 糖基化 | 50 |
泛素化 | 225 | 低氧缺氧 | 50 |
circRNA | 221 | 相分离 | 50 |
lncRNA | 204 | 泛凋亡PANoptosis | 42 |
细胞焦亡 | 175 | 细胞衰老 | 37 |
组蛋白 | 171 | 胞葬 | 33 |
肠道菌群 | 133 | CRISPR | 33 |
乙酰化 | 125 | 增强子 | 29 |
内质网 | 125 | 精氨酸甲基化 | 25 |
转录调控 | 112 | 迁移体 | 8 |
糖酵解 | 112 | 血管拟态 | 8 |
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。





























 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)