
2023-06-26 来源 : 芒果师兄聊生信

这篇综述题目为The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth,从免疫微环境角度总结了肿瘤起始到转移性生长的最新进展,全面整理了很多肿瘤的表型。非常经典!
本次翻译由朱医生独自完成,非常感谢朱医生百忙之中的付出和贡献

癌症归属复杂的生态系统,包括肿瘤细胞和大量的非癌症细胞,其嵌入在动态变化的细胞外基质中。肿瘤微环境(TME)包含各种免疫细胞、癌症相关成纤维细胞、内皮细胞、周细胞以及各种组织固有细胞。这些宿主细胞曾被认为是肿瘤发生的旁观者,现今观点认为在肿瘤发生、发展中发挥着重要作用。TME细胞成分与功能状态取决于肿瘤发生的器官、癌细胞内在特征,肿瘤发生阶段以及患者自身特性。作者从TME肿瘤发生、进展、侵袭和内渗到传播和远处生长的重要性进行综述。了解疾病进展的细胞内、外因素以及系统介质对于有效抗肿瘤治疗的发展起到关键作用。
引言
在过去几十年,我们对于肿瘤的理解有了本质上地变化。目前,癌症被看作不仅仅是一种遗传疾病,而且是一个复杂的生态系统疾病,涉及广泛的非癌症细胞及其在肿瘤内的相互作用。了解基因的改变是必要的,但是对于认识肿瘤的发生、发展还不足够。通过实体瘤镜检,癌症复杂性变得显而易见,揭示TME是一个高度结构化的生态系统,包含被多种非恶性细胞类型包围的癌细胞,这些细胞共同嵌入改变的、血管化的细胞外基质(图 1)。
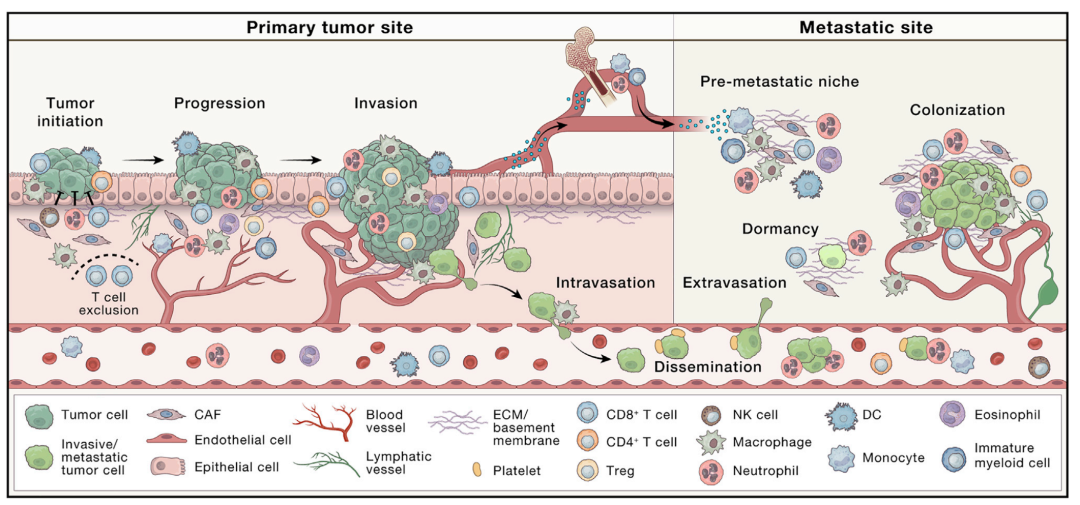
图1. 微环境调控原发肿瘤的进展、转移
TME包含各种免疫细胞、癌症相关成纤维细胞、内皮细胞、周边细胞和其他因组织而异的细胞类型,如脂肪细胞和神经元(表1)。最初,这些宿主细胞被视为肿瘤发生的旁观者。然而,作为机制研究结果,包括在预临床试验肿瘤模型中,现在TME细胞及其分泌的分子被认为在癌症的发病机制中有重要作用,因此成为有潜在价值的治疗靶点。TME细胞组成和功能状态会有所不同,取决于肿瘤发生器官、癌细胞的内在特征、肿瘤发生阶段和患者自身特性,TME中的各种细胞可以是起肿瘤抑制或肿瘤支持作用。在讨论TME 特性的广泛交织和对立的过程前,我们将概述 TME 在肿瘤发生的不同阶段形成和动态演化其中包括异型细胞相互联系的基本原则和文章可信程度的重要性。


癌细胞与宿主细胞之间的相互交流
癌细胞通过召集、重组非癌性宿主细胞以及重塑脉管系统和细胞外基质 (ECM) 以构建肿瘤支持环境。这个动态过程取决于癌细胞与 TME 的原驻或所召集非癌细胞间的异质作用。在近期计算机分析与建模的进展中,使用单细胞转录组学数据、大量肿瘤表达图谱和空间转录组学2-4揭示了TME中细胞间信号网络的多样性。这些图谱作为强大的假设生成数据集来指导后续的功能研究,这些研究揭示了复杂的细胞间相互作用是如何整合的,从而导致 TME形成和进化。这种细胞间对话通过多种机制进行调节,包括细胞间接触和旁分泌信号转导5。可以依赖接触性粘附分子介导,包括整合素、钙粘素、选择素和免疫球蛋白超家族成员,也可以通过缝隙连接和隧道纳米管介导。例如,癌细胞上的异常聚糖唾液酸化调控多数相互作用,包括伴Siglec表达的免疫细胞,促进免疫逃避和肿瘤进展6(图2)。众所周知, TME中接触依赖性细胞间信号传导的另一个的例子是 PD-L1/PD-1 通路(图2)。与相关骨髓细胞的癌细胞经常过度表达免疫检查蛋白PD-L1,该蛋白与适应性免疫细胞上PD-1受体结合以抑制免疫监视。这说明TME 通信分子敏感性程度具有关键的治疗价值,因为通过免疫检查点阻断 (ICB) 抑制 PD-L1/PD-1已成为越来越多癌症的标准治疗7。
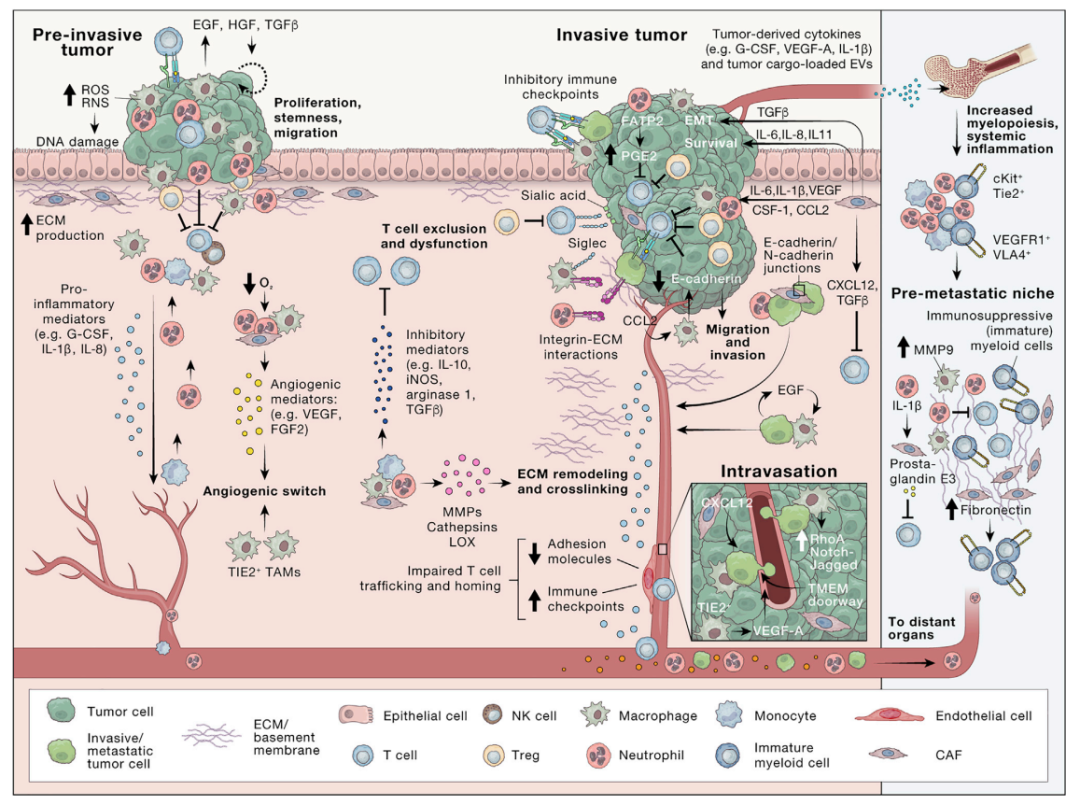
图2. 原发性肿瘤进展和TME内复杂的相互作用
除了直接细胞间接触外,细胞因子、趋化因子、生长因子和蛋白酶旁分泌信号对于TME内的细胞间通讯也至关重要。在癌症内在特征和细胞应激下分泌的这些分子,它们可以源自 TME中的多种细胞类型,并通过与其受体结合或通过 ECM 重塑对靶细胞发挥直接和间接作用。包括外泌体在内的细胞外囊泡 (EV) 的释放是另一种旁分泌机制,它可以改变局部环境8,甚至对原发肿瘤部位产生深远影响9(图1)。临床前试验研究表明,黑色素瘤衍生的 EV 可引导骨髓 (BM) 原始细胞形成促血管生成表型,从而促进转移形成10。癌症衍生表达 PD-L1的EV可以抑制引流淋巴结中的 T 细胞活化,从而促进肿瘤进展和 ICB 耐药。 11,12改变代谢需求和伴随代谢物分泌在创造支持性 TME 中的重要性也越来越被认可。 13与体内实验结合的单细胞代谢组学和空间多组学14,有望提高我们对 TME 中代谢物串扰和竞争的理解。

ECM 通过充当隔离分泌分子的储库以及细胞粘附和迁移的底物来促进细胞间联系。蛋白酶解离束缚的分子对 ECM 进行重塑,从而产生局部高浓度的释放介质。此外,癌症和 TME 细胞通过受体(包括整合素和 CD44)直接接触周围的 ECM,有助于在癌症中发挥作用的复杂信号网络 15(图 2)。综上,肿瘤细胞和非肿瘤细胞之间的联系可以通过多种机制在多个层面发生。此综述将讨论说明沿肿瘤发生轨迹的细胞间 TME 串扰机制的代表性示例。
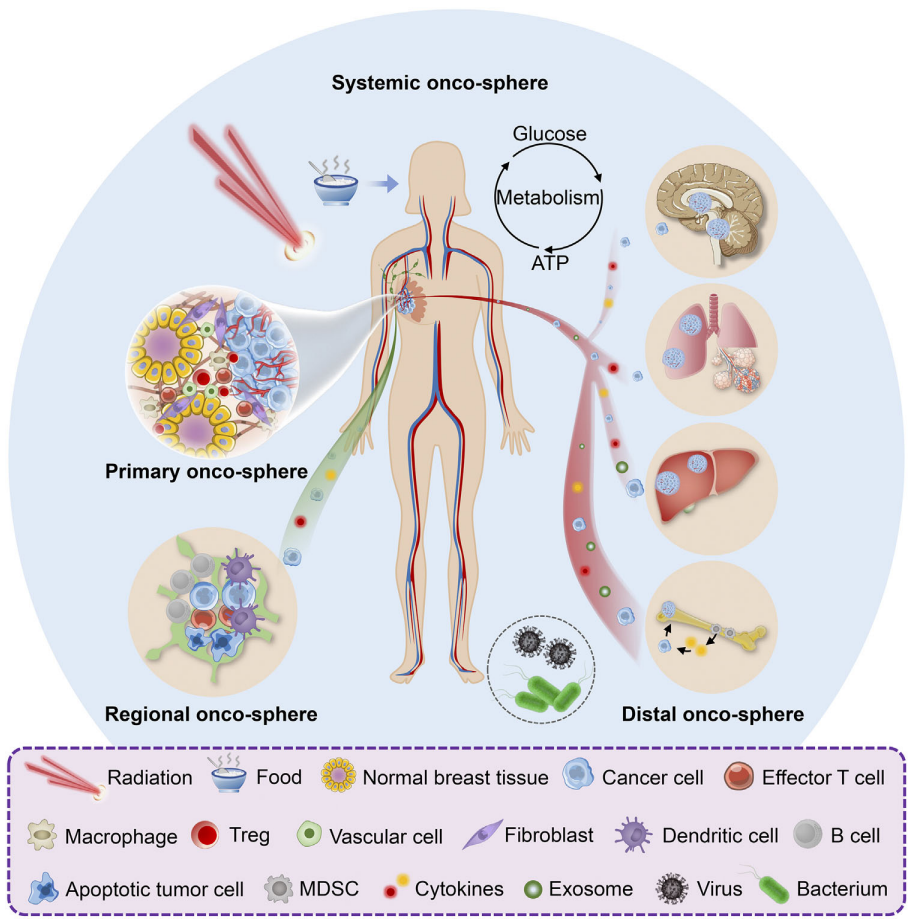
TME组成和功能状态在不同患者间可能存在很大差异,即使在同一癌症类型中也是如此。 16-18包括年龄、性别、生活方式、体重指数和微生物组在内的个体差异因素会影响 TME,肿瘤发生器官也会影响 TME。不同的器官具有独特的组织固有免疫和基质细胞类型,组织类型可以决定这些细胞的功能状态。说明性的例子包括在不同器官中发现的功能不同的巨噬细胞群。 19例如,肝脏中的固有巨噬细胞称为枯否细胞,在转录组和生理功能方面与肺中的肺泡巨噬细胞或大脑中的小神经胶质细胞不同。目前,前沿技术也揭示了细胞表型、激活状态和命运对其他免疫和基质细胞类型(包括中性粒细胞、成纤维细胞、T 细胞、EC 和脂肪细胞)的组织环境的适应。 20–25这些细胞的器官特异性转录程序可能在它们到达组织时由局部线索激发,或者可能在组织发育过程中已经被表观遗传印记,如长寿成纤维细胞的情况。 26人们越来越认识到,稳态条件下细胞的器官特异性印记可以部分解释这些细胞在不同肿瘤类型中的不同表型和功能。 27,28例如,在人类和小鼠非小细胞肺癌中,组织固有巨噬细胞与单核细胞来源的巨噬细胞相比具有明显的时空分布和功能。 29随着器官间细胞编程的差异,ECM中基质蛋白的组成也各不相同。 30鉴于ECM在调节细胞表型和行为方面的重要性,这种组织依赖性 ECM 特性有助于产生器官特异性的TME。
突变和肿瘤,谁是因,谁是果,值得探索!
除了解剖学位点依赖机制外,TME 最重要的调节因子是癌细胞本身。大脑中出现的神经胶质瘤与源自颅外肿瘤的脑转移具有不同的免疫环境,这一发现支持了这一点。18,31逐渐明了的是癌细胞内在特征,包括遗传学改变、代谢重译和信号失调,是肿瘤塑造其微环境的关键决定因素。临床前试验表明,操控癌细胞内在编译会改变分泌蛋白组,改变细胞表面受体或配体,影响 EV 的运载量和浓度,并改变营养物质的使用,从而导致肿瘤免疫环境的广泛变化和 ICB 反应的障碍。 32,33例如,KRASG12 驱动肺腺瘤中的 MYC激活导致上皮细胞分泌 CCL9 和白细胞介素 23 (IL-23),形成炎症、血管增生和免疫抑制状态的TME,从而增强了肿瘤发生。 34在黑色素瘤中,肿瘤固有信号 β-连环蛋白诱导转录抑制因子 ATF3 的表达,从而抑制 CCL4分泌。这损害了 CD103+ 树突状细胞 (DC) 的聚集,导致 T 细胞排斥和 ICB拮抗。 35 最后,癌细胞中 Trp53 的突变或缺失导致富含骨髓的免疫抑制环境,从而促进肿瘤进展、36,37 转移、38 和 ICB 拮抗。39随着高分辨率分析技术的快速发展,将揭示癌症内在特征与 TME 之间的更多联系,根据因果实验证明,这可能为针对个体肿瘤定制的、合理的 TME 靶向策略奠定基础。
肿瘤起始:组织稳态的瓦解
恶性细胞必须克服多个瓶颈才能形成肿瘤,这些瓶颈则取决于阻碍周围组织的正常信号,然后劫持微环境以供给发展中的肿瘤。本节主要阐释新生肿瘤如何逃避免疫攻击,如何将周围基质转化为支持肿瘤生长的 TME,以及它们如何获得足够的氧气和营养供应以满足其高代谢需求。
肿瘤起始——从免疫攻击到免疫逃避的平衡
免疫系统在抵御威胁生命的病原体、伤口愈合和清除受损细胞都有至关重要的作用。为了执行这些功能,免疫系统具有不可思议的多样性和适应性,其严格的调控机制来减轻组织损伤和恢复体内稳态。然而,尽管适应性免疫细胞具有识别和消除病原体及表达非自身抗原的细胞的能力,但癌细胞可以逃避攻击并发展成成熟的肿瘤(图 1)。
研究发现,在已形成的肿瘤中检测到对癌细胞表达新抗原具有特异性的 T 细胞,40 并且高 T 细胞密度、 T 细胞活化特征与提高各种癌症类型的存活率相关,41,42表明适应性免疫系统具有识别癌细胞的潜力。事实上,临床预试验使用高免疫原性肿瘤模型为癌症免疫监视学说提供了早期实验证据,以假设适应性免疫系统可以抑制和塑造肿瘤。 43,44尽管如此,许多发展中的癌症已经成功地避免或抵抗免疫攻击,这在肿瘤发生的早期就已经存在。例如,虽然适应性免疫系统受到抑制的患者(如艾滋病患者或受过器官移植的患者)患病毒相关恶性肿瘤的风险较高,但非病毒相关上皮癌的发病率并未增加。 45同样,在各种转基因小鼠模型中,适应性免疫细胞成分的基因消除后肿瘤发病率并非都是增加,反而会出现降低的情况。 46–49这些发现强调需要更好地了解驱动癌症免疫逃避的机制。
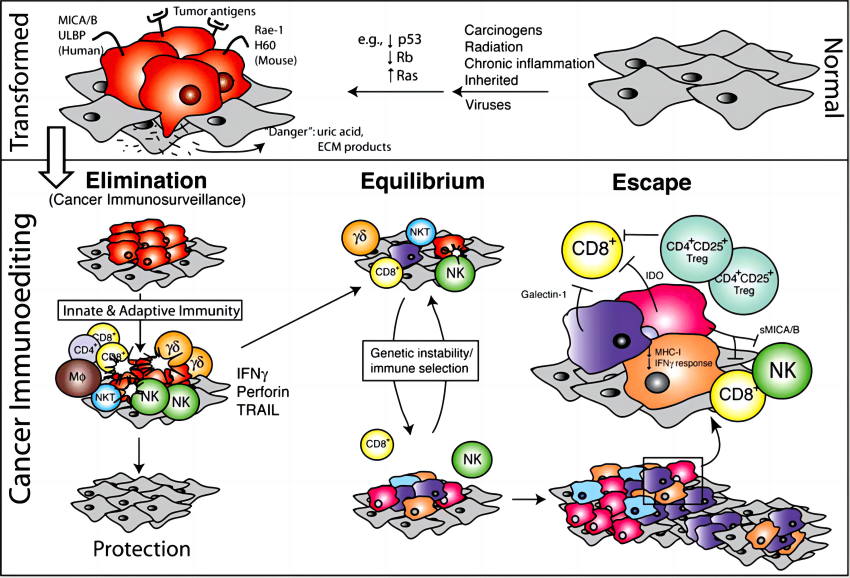
近期研究利用单细胞技术和多重空间分析,揭示了癌前早期步骤以及免疫环境中共同进化的空间、分子、结构和功能变化。 50肺癌的演变分析表明,最初低度病变的特征是大量涌入幼稚 T 细胞,表明免疫系统在其最早阶段就感知到了这种病变。然而,随着病变的进展,观察到活化 T 细胞和髓样细胞的积累转变,以及免疫抑制相关基因的正向调控。 51同样,对比乳腺导管原位癌 (DCIS) 与正常乳腺组织中的免疫细胞组成,发现 DCIS 中的白细胞总数更多、中性粒细胞更多,CD8/CD4 比例降低。DCIS 向浸润性导管癌的进展伴随着过渡到抑制的免疫环境,标志活化的 CD8+ T 细胞减少、PD-L1 和 CTLA4 表达增加、调节性 T 细胞 (Treg) 增加以及克隆型 T 细胞受体 (TCR) 多样性降低。52在头颈癌患者中,早期病变与引流淋巴结中的免疫刺激性中性粒细胞相关,在后期发展为免疫抑制性嗜中性粒细胞。 53总而言之,由这些患者的分析表明,适应性和先天性(固有)免疫系统可以感知早期肿瘤病变,并且随着这些病变的进展,而后是向免疫抑制 TME 的转变。
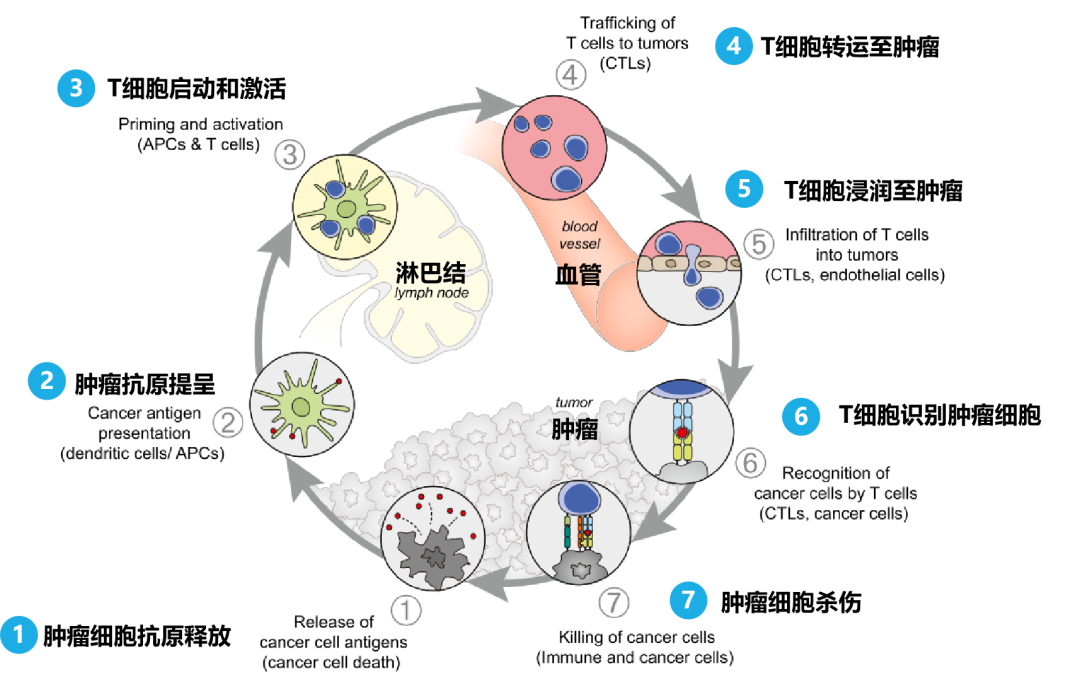
临床预实验肿瘤模型进一步支持肿瘤进展过程中免疫逃避的早期发生。在转基因乳腺癌模型中 Brca1 和 p53 功能的丧失驱动的乳腺肿瘤恶变前阶段的时间分辨单细胞分析揭示了潜在免疫抑制环境的早期建立,其特征是 Treg 和组织固有巨噬细胞的积累。54同样,胰腺细胞中具有Kras 突变的小鼠的早期肿瘤进展通过细胞警报因子 IL-3355 伴随的组织损伤而加速。用重组 IL-33 足以引起染色质失调并加速 Kras 突变推进胰腺上皮内瘤变 (Pan-IN) 的发展,说明基因-环境相互作用以触发癌症背后的基因调控程序。 55肿瘤起始期间免疫逃逸的时间和机制可能取决于组织环境、肿瘤起始基因改变和宿主特征。几种癌症的出现伴随着慢性炎症的发生,从而利用有利于免疫抑制的已经被破坏的骨髓适应性免疫细胞进行串扰。慢性发炎组织通常伴随 Th2 型免疫反应和骨髓细胞的积累,这些细胞极化朝着免疫抑制功能状态进行,分泌活性氧 (ROS)、促炎细胞因子、趋化因子、生长因子和促血管生成介质。这些可能共同导致组织损伤、上皮突变、内皮功能障碍和血管增生、免疫抑制和基质重塑,最终导致肿瘤的发生和发展。56,57众所周知,引发肿瘤的炎症性疾病的例子包括慢性肠炎,它易患结直肠癌;慢性肝炎和非酒精性脂肪肝是诱发肝癌的基础;石棉引起的炎症,可导致间皮瘤。 57肥胖引起的慢性炎症也会增加患不同癌症类型的风险,包括乳腺癌和子宫癌。 58–60
无论癌症是由于长期慢性炎症而发展,还是肿瘤在发生早期阶段调控支持肿瘤生长的炎症环境,几乎所有进展中的肿瘤都会诱导不同水平的 T 细胞、NK细胞和 DC 排斥或 CD8+ T 细胞功能障碍的触发程序。 61–65当肿瘤同时刺激骨髓细胞(特别是巨噬细胞和中性粒)的聚集和激活时,就会发生这种情况,它们共同形成肿瘤支持性炎症环境。
炎症——肿瘤发展的催化剂
癌症引起的炎症与伤口中观察到的炎症反应进行比较。 66 然而,虽然伤口愈合是适应性和先天性免疫细胞之间动态配合的相互作用导致炎症消退和组织稳态恢复,但在发展中的肿瘤是被破坏的适应性先天免疫细胞串扰从而无法解决。在延长炎症信号的影响下,缺氧、低 pH 值和代谢物水平改变,使得炎症具有慢性和破坏性。因此,Dvorak将肿瘤比作无法愈合的伤口。67在本节中,我们讨论了癌症相关炎症的几个关键特征。由于控制肿瘤相关免疫细胞的组成、空间组织和激活状态的机制多种多样,并且肿瘤之间差异很大,因此重要的是要考虑炎症的类型及其对癌症进展的影响将取决于肿瘤类型和不同患者。

随着肿瘤的生长,由于细胞毒性 CD8+ T 细胞和 NK 细胞的逐渐减少、功能失调的 CD8+ T 细胞、免疫抑制性 CD4+FoxP3+ Treg 和调节性 B 细胞的增加,共同进化的免疫环境发生了深刻的变化,而 CD4+ T 细胞是 倾向于促炎 Th2 表型,DC 表现出成熟和功能的缺陷(表 1)。与此同时,骨髓细胞越来越多地动员到 TME,在那里它们调整自己的表型以适应局部炎症信号。肿瘤相关巨噬细胞 (TAMs) 和中性粒细胞 (TANs) 通常是不同 TME 中最丰富的骨髓细胞,并已得到广泛研究。68–70驱动这些细胞动员和激活的关键肿瘤衍生介质包括 CSF-1、CCL2、VEGF-A、肿瘤坏死因子 α (TNF-α) 和作用于巨噬细胞的的信号蛋白 3A ,G-CSF、GM-CSF、IL-6、CXCL1、CXCL2、IL-1b 和作用于中性粒细胞的IL-8。68它们在人类肿瘤中的存在通常与较差的预后和较差的治疗反应相关,尽管在某些情况下,它们的存在与良好的结果相关。 42,68–70最近的研究揭示了肿瘤相关骨髓细胞巨大的多样性和可塑性。从初始分类到经典与替代激活的简单二元状态,即 M1 和 M2 巨噬细胞 71 或 N1 和 N2 中性粒细胞 72,(单细胞)转录组学分析研究和功能分析提供了重要的新见解。73,74例如,单细胞 RNA 测序 (scRNA-seq) 分析表明多个巨噬细胞和中性粒细胞亚群在单个肿瘤中共存,表明 TAM 同时共表达典型的 M1 和 M2 标记基因。74,75目前尚不清楚这些亚型是代表不同的人群还是同一人群的不同状态。然而,越来越多的研究支持这样的观点,即这些不同的骨髓细胞簇显示出截然不同的功能,有时甚至是相反的功能。73例如,TAN 经常发挥免疫抑制功能,但一个特有的 TAN 亚型被证明在早期人类肺部肿瘤中具有抗原呈递能力。76在 TAM 群体中,还鉴别了具有免疫抑制或促血管生成特征的亚群。70最近的一项泛癌分析表明,巨噬细胞亚群在不同肿瘤类型之间表现出不同的转录组学模式,74这就支持肿瘤相关骨髓细胞的器官和癌症类型特异性印记的概念。在肿瘤局部和整体,了解癌症中骨髓细胞亚群的全谱有助于设计方法以治疗性利用具有抗癌特性的骨髓免疫亚群,同时抑制或耗尽具有肿瘤支持作用的免疫亚群。
关于进化肿瘤中巨噬细胞多样性的另一层复杂性是组织固有巨噬细胞,它们由胚胎源性巨噬细胞在各个器官中起源,在功能上与聚集的单核细胞衍生巨噬细胞不同。19,68虽然尚未描述中性粒细胞在个体发育方面类似的差异,但临床预实验表明,来自发展中肿瘤的炎症介质重新编程 BM 造血,使其倾向骨髓系并改变 BM 的嗜中性粒细胞输出。77随后以组织和肿瘤特殊的方式对它们的命运和行为进行额外的转录和表观遗传适应。21最有可能的是,其他骨髓细胞(包括嗜酸性粒细胞、肥大细胞、嗜碱性粒细胞和 DCs)经历了类似的多层次肿瘤诱导教育过程,其程度可能由不同骨髓细胞亚群的不同寿命和更替决定。大量的 TAM 和 TAN与不良患者的预后之间存在关联一致,42,68-70 在小鼠模型中这些细胞的耗竭、抑制或重组会损害许多癌症的发展1,58,59,78–83并且还提高了化学疗法、放射疗法和免疫疗法的疗效。1,84–89然而,在一些临床前期,巨噬细胞或中性粒细胞的联合效应起到抗肿瘤作用。69,90–94 这些肿瘤抑制特性是否由某些癌症(亚)类型、阶段或其他肿瘤或宿主相关特征引起,还需要深入研究。肿瘤相关骨髓细胞显示出高可塑性,可以影响许多致瘤过程,包括 (1) 通过癌细胞的增殖、存活和侵袭能力直接调节癌细胞的命运和行为,(2) 产生免疫抑制的 TME,(3) 激活肿瘤血管生成,以及 (4) 重塑 ECM。
我们对在不同肿瘤中发挥作用的机制的理解仍然有限,不同的机制可能共存于同一肿瘤中,可能局限于肿瘤内的特定空间区域,或者可能随着癌症的进展而依次被激活。我们在这里所强调几个例子,说明 TME 中骨髓细胞效应机制丰富多样,并建议读者参考最近的综述以进行详细讨论。 68,69,87,95
对于长期激活的巨噬细胞和中性粒细胞如何直接促进上皮细胞恶性转化的一个重要机制是通过产生活性氧和氮物质,这些物质可以直接诱导上皮细胞中的 DNA 损伤。56,57肿瘤相关骨髓细胞还分泌丰富的生长因子和细胞因子,改变癌细胞的命运和行为,包括维持癌细胞增殖和迁移的表皮生长因子 (EGF)96;肝细胞生长因子 (HGF),可增加癌细胞的转移潜能97且转化生长因子 β (TGF-β)、IL-6 和 IL10 以及 GPNMB,它们支持癌细胞干性。 98–101 然而,在某些情况下,例如在某些早期肿瘤或基于抗体的治疗期间, 骨髓细胞也可以杀死或吞噬癌细胞,或促进抗体依赖性细胞的细胞毒性,76,92,95 可以看出这些细胞在 TME 中相反的作用。
除了直接影响癌细胞外,TAM、单核细胞原始细胞、中性粒细胞和研究较少的肥大细胞还可以通过调控 TME 中的支持作用间接促进肿瘤发生。来自临床预实验的数据可知,骨髓细胞具有强大免疫抑制作用,表明 TAM 或 TAN 的耗竭或功能重编程可减少肿瘤浸润性 T 细胞的免疫耗竭程序,恢复抗肿瘤免疫反应,并与 ICB 疗法具有协同作用。 81,87,89,102此外,TAM 和 TAN 是否充裕通常与患者的 ICB 反应差相关。 103
骨髓细胞采用多种机制为肿瘤进行免疫逃避。它们可以分泌T细胞和NK细胞的抑制性介质,包括IL-10、ROS、iNOS、精氨酸酶1、TGF-β,表达PD-L1等免疫检查点分子immune checkpoint molecules,产生炎症介质IL-1β、TNF-α, 和 IL-6 以放大炎症反应。 68,87,104,105例如,在早期人类肺癌中,与健康肺中的巨噬细胞相比,浸润性巨噬细胞亚群具有高水平 PPARγ、CD86下降和PD-L1 增加,这与 T 和 NK 细胞存在减少相关。 65泛癌 scRNA-seq 分析确定了 IL-4I1+ PD-L1+ IDO1+ TAM 亚群与 T 细胞耗竭、色氨酸降解和 Treg 积累有关。 106由于它们的 RNA 含量低,TAN 在 scRNA-seq 数据代表性较低。但是,有充分的临床和实验证据表明它们在肿瘤局部和整体具有强大的免疫抑制能力。 108事实上,由于这种能力,这些中性粒细胞和单核细胞通常被归类为统一的术语——髓源性抑制细胞 (MDSC)。105,109 由于这些细胞的免疫抑制能力只是它们可以发挥的不同肿瘤支持功能的一个方面,所以我们在这里仍使用传统命名法。
通过它们在 TME 中的代谢适应和营养物质以及必需氨基酸的消耗,骨髓细胞促进癌症免疫逃避和血管生成成为一种新兴机制。肿瘤相关骨髓细胞通常改变糖酵解活性、增加谷氨酰胺和脂肪酸消耗,这可以通过营养和免疫逃避机制支持肿瘤生长。110,111例如,肿瘤诱导的中性粒细胞中脂肪酸转运蛋白 2 (FATP2) 的上调使花生四烯酸加工成前列腺素 E2,从而增强其促肿瘤、免疫抑制特性。112骨髓细胞还可以通过耗尽 TME 中的胱氨酸和半胱氨酸来抑制 T 细胞活化。有关 TME 中骨髓细胞代谢可塑性的详细概述,我们建议读者参考最近的综述。 110,111,114除了调节免疫抑制外,骨髓细胞在肿瘤发生和发展中协调 ECM 重塑和血管生成。以上过程是如何被肿瘤相关免疫细胞促进,将在下文讨论。
CAF 和 ECM 重塑在发展中TME的多方面作用
CAF 与免疫细胞一起构成许多肿瘤的主要成分。115,116一些肿瘤,例如肝癌,是由于成纤维细胞异常活化而发展起来的,特别是在纤维化或肝硬化的肝脏中。117 其他类型的癌症也可以诱发纤维化,通常在其发生和进展期间被称为结缔组织增生。 118单细胞技术的最新进展揭示了 CAF 以前未被重视的表型和功能多样性。 116 例如,人胰腺癌(PDAC)早期病变的 scRNA-seq 揭示了肿瘤起始期间 CAF 亚群的组成和转录组的动态变化。119 Barrett食管进展为食管腺癌的特征是基质和成纤维细胞炎症相关基因表达增加。120同样,在多种小鼠模型中,CAF 组成和转录组的改变是肿瘤发展过程中的早期标志,这些变化随着肿瘤的发展而演变。121-123
肿瘤中CAF 的起源仍然存在争议,并且可能因肿瘤分期和癌症类型而异。局部组织固有成纤维细胞的扩增可以代表早期肿瘤中 CAF 的来源。 115,124其他研究表明,一些组织具有不同的成纤维细胞系,125,126 这可能导致不同的细胞状态或功能多样的 CAF 亚群。126 CAF 也可能来自其他细胞类型的转化,包括肌成纤维细胞、骨髓间充质干细胞 (MSC)、星状细胞和脂肪细胞衍生的 CAFs。127–130这些不同起源的CAF导致其表型和功能异质性。
CAF 还在响应来自 TME 的动态变化的线索上表现出可塑性。这种可塑性的程度尚不完全清楚,但最近的研究表明,CAF 由多种亚型组成,这些亚型在肿瘤进展过程中发生变化,并且在空间上受到调节。 115,131在胰腺癌中,三种不同的 CAF 亚型共存:肌成纤维细胞 (myCAFs)、炎性 CAFs (iCAFs) 和抗原呈递 CAFs (apCAFs),它们具有不同的功能特性和转录组学可塑性。131-133在其他癌症类型中,已经确定类似但也有其它CAF亚群。131,134-136 有趣的是,CAF簇分布可以随着中断性机械信号传导或免疫治疗后发生变化,137 进而提出了调整 CAF 亚群组成的策略。
CAF 被 TME 中的各种机制激活,包括暴露于炎症介质、ECM 硬度和成分的变化以及代谢物的改变。115关键的激活剂包括 TGFb、IL-1、IL-6 和 TNFa,115 它们也会在发展肿瘤环境下中驱动慢性炎症,强调肿瘤发生和进展期间炎症和 CAF 之间的联系,下文进一步讨论。基质硬度通过刺激 CAF 中的 YAP 和 MRTF-SRF 调节网络促进转录重新连接,从而驱动促纤维化反应、ECM 蛋白的产生、血管生成和癌细胞侵袭。138,139肿瘤衍生信号还可以调节 CAF 中的复杂信号网络。例如,在 PDAC 中,癌细胞以旁分泌方式激活 CAF 中的 Hedgehog 信号。140 早期临床预实验表明,靶向 Hedgehog 通路可使 PDAC 肿瘤对吉西他滨敏感。 141然而,Hedgehog 通路抑制剂联合化疗的临床试验并未显示任何治疗益处,在某些情况下甚至加速了肿瘤进展。142–144 现在人们认识到 Hedgehog 信号在 myCAF 和 iCAF 中的激活差异。因此,Hedgehog 通路抑制减少了 myCAFs 并增加了 iCAFs,导致更多的免疫抑制的 TME。 143与肿瘤相关免疫细胞相似,CAF 在 TME 中同样发挥多效性和功能相反的功能。 145 CAF 促进肿瘤功能的早期证据来自在小鼠中将癌细胞与 CAF 共同注射的实验。 123进一步的临床预实验,其中内源性 CAF 在基因或治疗上设为靶向,另外揭示了 CAF 强大的致癌性。131,146–148 然而,特定 CAF 亚群(包括肌成纤维细胞)的消耗或靶向在某些小鼠模型中加速了肿瘤生长,149,150 在不同的 TME 中涉及功能相反的 CAF 亚群。
CAF 主要负责 TME 内的 ECM 沉积和重塑。151 例如,TME 中的纤维化导致组织僵硬,这与胰腺癌和乳腺癌患者的生存率低下显着相关。152ECM 的机械特性直接影响癌细胞的信号传导和行为,此外还影响免疫细胞的聚集和激活,并减少药物进入肿瘤。此外,在这种情况下,CAF 和免疫细胞协同工作。纤维化肿瘤具有发炎表型,炎症促进纤维化。153-155骨髓细胞是 ECM 重塑酶、基质金属蛋白酶 (MMP) 和组织蛋白酶以及胶原交联酶(包括赖氨酰氧化酶 (LOX))的重要来源。在多个临床预实验中可以证明其促进肿瘤发生、侵袭和耐药。 153,156–159
最近的研究表明,CAF 可以通过多种机制帮助肿瘤逃避免疫控制。在人类癌症中,CAF 与 T 细胞功能障碍和排斥有关,前临床研究表明,CAF分泌 CXCL12 和 TGF-b 或以 ECM 沉积形成物理屏障直接阻止 T 细胞聚集或激活。 134,160–162在多数患者中,CAF 诱导的 T 细胞排斥可能发生在肿瘤早期,因为 MYH11+ αSMA+ CAF 在一些早期 NSCLC 病变中在肿瘤巢周围形成单层细胞,其与巢内 T 细胞密度降低相关。 134有趣的是,在 PDAC 中,表达主要组织相容性复合物 (MHC) II 类 CAF 的亚群显示出与 CD4+ T 细胞相似的抗原呈递能力,但缺乏共刺激分子,这可能会导致 T 细胞活化缺陷,131,132 从而赋予另一层免疫调节功能 在 CAF 上。CAF 还通过分泌 IL-6、IL-1b、VEGF、CSF-1、CCL2 和几丁质酶 3-like1 等介质动员和编程免疫抑制性骨髓细胞,并通过促进积累和免疫抑制活性,间接干扰抗肿瘤免疫调节性T细胞 (Tregs)。163-167CAF 的免疫调节特性为逆转免疫抑制和改善 ICB 治疗提供了机会。事实上,多项前临床研究表明,CAF 调节后会加强 T 细胞流入和 ICB 疗效。 160,168,169对 CAF 与免疫细胞之间相互作用机制,以及患者和癌症类型之间的异质性需进一步理解,可能会激发作用于逆转 CAF 诱导的免疫抑制同时刺激 T 细胞功能的新型联合疗法。
CAF 还直接影响癌细胞。在人类乳腺癌和肺癌样本中,CD10+GPR77+ CAF 亚群通过 IL-6 和 IL-8 分泌为癌症干细胞提供生存环境,从而促进肿瘤形成和化疗耐药。170在结直肠癌中,CAF通过 TGF-β 驱动的 IL-11 分泌,促进依赖性GP130/STAT3癌细胞生存程序地扩散。 171胰腺星状细胞中胰腺肿瘤发生诱导的脂质代谢转变导致溶血磷脂酰胆碱分泌,从而支持 PDAC 细胞增殖和迁移以及 AKT 激活。 172这些有代表性的例子说明,CAF 改变癌细胞信号和行为的机制具有多样性和组织依赖性。
综上,CAF 一系列功能具有高度多样化且依赖于环境。在单细胞测序和多组学方法的最新进展上,以及复杂的谱系追踪模型和增进对定义 CAF 亚群的环境功能特性的理解,将促进对促肿瘤CAF 亚群的精细靶向策略的开发。
血管生成促进癌症进展
血管生成是新血管形成的过程,对于生长至关重要(图 1)。一旦肿瘤生长超过 1-2 毫米,就必须建立自己的氧气和营养血管供应。 173在对看似健康的个体进行尸检研究中,在包括乳腺、前列腺和甲状腺在内的几个器官中检测到微小静止肿瘤,其患病率远高于根据报告的这些组织中癌症发病率所预期的患病率。 174-177血管生成的缺乏被认为是为什么一些微观病变不会发展成浸润性癌症而是保持休眠状态的原因。 177在健康组织中,脉管系统是稳定的,而作为血管主要组成部分的 ECs 并没有积极分裂。相比之下,肿瘤发生是血管生成的开始,也称为血管生成开关,178是一个复杂的过程,涉及 EC、周细胞、壁细胞、癌细胞、肿瘤相关免疫细胞和 CAF 之间的广泛串扰。179,180已存在血管中新生毛细血管萌芽的脉管系统物理变化,以及 EC 显着的异质性和可塑性已详细讨论。 25,181-183 肿瘤血管不断暴露促血管生成物质,导致组织混乱、渗漏 和曲折的脉管系统,伴随周细胞覆盖缺陷和 EC 间断被覆。这会影响肿瘤的氧合作用,改变免疫细胞动力学,并减少药物对肿瘤的渗透。 179,180 一种可替代的血管形成过程涉及血管共同选择,其中肿瘤扩张先前存在的血管,而不需要刺激新的血管生成。 184癌细胞可以沿着宿主血管的近腔表面迁移,并且这些血管可以并入正在发育的肿瘤中。185 血管拟态代表了另一种使进展中的肿瘤进入循环的方法,这已在黑色素瘤和胶质母细胞瘤中得到报道。这涉及形成与现有脉管系统相连的癌细胞通道、有无基质蛋白沉积。 186 这些过程背后的机制尚未完全了解,但它们可能使肿瘤阻碍抗血管生成疗法。184
缺氧,即组织中缺氧,是血管生成的主要诱因。许多在缺氧条件下活跃的分子可以促进血管生成转换,其中血管内皮生长因子(VEGF)及其下游信号通路是主要驱动因素。在许多患者中,高肿瘤内和全身 VEGF 水平与各种癌症类型的不良疾病结果相关。 187,188抑制 VEGF 信号可以阻止小鼠的血管生成和肿瘤生长,189,190 表明血管生成是肿瘤发生的关键步骤。其他促进血管生成的分子,如碱性成纤维细胞生长因子 (FGF2) 和胎盘生长因子 (PIGF),也存在于肿瘤中,以及炎症介质,包括 TNF、BV8 和 G-CSF。179肿瘤进化过程中血管网络的形成和持续适应受癌细胞和宿主细胞的调节,这种调节方式与环境相关。180 在本节中,我们着重讨论骨髓细胞和 CAF 作为肿瘤血管生成的原型驱动因素。为了进一步讨论其他肿瘤相关宿主细胞如何调节血管生成,我们建议读者参阅综合综述。 179,180肿瘤相关骨髓细胞通过促血管生成介质(包括 VEGF-A、FGF2、PIGF、TNF 和 BV8)促进肿瘤血管生成和增加血管通透性。这些细胞还产生蛋白酶,例如 MMP 和组织蛋白酶,它们分解 ECM 并释放隔离的促血管生成分子,使它们具有生物可利用性。 180,191越来越多的证据表明,特定的骨髓细胞亚群具有促血管生成功能。73 例如,在胰岛癌发生模型中,产生 MMP-9 和 BV8 的中性粒细胞驱动血管生成。192,193 表达 TIE-2 的巨噬细胞存在于 肿瘤的血管周围微环境 (PVN),在不同的小鼠肿瘤模型中驱动血管生成。 194-196 经历了代谢变化的缺氧 TAM 也通过与 EC 竞争葡萄糖,促进了无组织、不稳定的肿瘤血管的形成。 197 对多种人类癌症中肿瘤相关骨髓细胞进行单细胞分析已经得出具有显著血管生成基因特征的亚群,尽管这些细胞的统一分子注释尚未建立。 73,74
重要的是,脉管系统和免疫细胞之间的相互作用是相互的。越来越多的证据表明,肿瘤诱导的血管生成有助于免疫抑制和免疫逃避。 198,199 例如,可以下调调节免疫细胞归巢和运输的血管粘附分子。肿瘤相关 EC 表达较低水平的 ICAM-1、VCAM-1、E-选择素和 P-选择素,这导致免疫细胞不能渗入肿瘤区域。 198,199相反,包括IDO、TIM3和PD-L1在内的抑制性免疫检查点分子可以在肿瘤血管上上调。198此外,据报道,EC诱导的肿瘤FasL表达可以选择性杀伤效应CD8+T细胞,导致免疫逃避。200单细胞研究揭示了对健康和疾病中不同EC亚群的免疫调节表型的新见解。199促血管生成介质也可以直接影响免疫细胞。例如,VEGF-A抑制DC的成熟,增加Tregs,并增强肿瘤相关骨髓细胞的免疫抑制状态。198,201–203最后,肿瘤血管、ECM和缺氧微环境的物理特性改变也会影响免疫细胞的浸润和功能。204
同样,肿瘤淋巴管也具有重要的免疫调节特性。205,206与血液内皮细胞一样,淋巴内皮细胞可以通过各种机制抑制T细胞反应,包括免疫检查点分子的表达和在缺乏共刺激分子的情况下的抗原呈递。205,206高水平的VEGF-C是淋巴管生成的主要驱动因素,与转移增加和存活率降低有关。207然而,矛盾的是,在黑色素瘤中,免疫疗法对具有高VEGF-C表达水平和显著淋巴管生成的肿瘤更有效。208了解血管生成和免疫抑制之间的肿瘤支持性互馈可能有助于利用抗血管生成疗法作为逆转免疫抑制和恢复抗肿瘤免疫的手段。事实上,在前临床研究中,破坏血管生成可以提高各种免疫疗法的有效性。209
综上,证明TME的复杂性、相互联系性,CAFs也是肿瘤血管生成的关键协调因子。与免疫细胞一样,CAFs产生几种促血管生成介质,包括VEGF-A、FGF2和CXCL12等。180,210此外,通过聚集和激活TME中具有促血管生成能力的EC原始细胞和髓系细胞,CAF间接促进肿瘤血管生成。此外,CAF介导的促结缔组织增生反应影响发展中肿瘤的血管形成。211 CAFs产生胶原交联酶,包括LOX和羟化酶,以及ECM降解蛋白酶,这些酶改变肿瘤的机械特性并影响血管生成。151然而,ECM蛋白水解后释放的一些胶原片段,如内皮抑素和肿瘤生长抑制素,可以抑制血管生成,211表明CAF依赖的促血管生成和抗血管生成过程在TME中都起作用。
肿瘤发展:为转移扩散奠定基础
癌细胞的侵袭和迁移
一旦肿瘤成功地在血管生成、炎症和纤维化之间建立了相辅相成的联系,它们就可以进入疾病进展的下一阶段:局部侵袭。侵袭性生长是癌症的主要特征之一,为转移性传播奠定了基础(图1和图2)。侵袭是一个复杂的多步骤过程,涉及癌症细胞彼此分离,从原发肿瘤块迁移,并侵袭周围的基质。212癌细胞可以作为单个细胞或以成串或成簇的方式进行侵袭。213侵袭期间,癌细胞暴露于不断变化的TME细胞和分子成分中,必须转换表型才能完成这一过程。为了与邻近的癌症细胞分离,必须破坏上皮细胞的粘附。细胞间粘附蛋白E-钙粘蛋白的缺失是这一过程的核心,通常伴有上皮-间充质(EMT)样过渡状态。癌细胞失去上皮特征,并获得茎状特性和迁移的间充质特征。214,215来自TME的物质促进癌细胞的表型转换,从而实现局部侵袭216,217(图2)。例如,在HER2+乳腺肿瘤的小鼠模型中,癌前病变中的上皮细胞和髓细胞产生的CCL2募集CD206+Tie2+巨噬细胞,其下调E-钙粘蛋白粘附并刺激Wnt信号传导。这导致了类似EMT的反应,有助于早期传播。218通过抑制CSF1R抑制巨噬细胞耗竭逆转了这一过程,导致E-cadherin在增生导管中的表达增加,并减少了癌症细胞的播散。218在非小细胞肺癌早期,癌症细胞定位于组织附近的肺泡巨噬细胞。从早期小鼠NSCLC病变中分离的肺泡巨噬细胞的转录组学分析显示,抗原呈递和组织重塑基因(包括蛋白酶)的表达增加。机制性体内外研究表明,组织固有巨噬细胞在邻近癌细胞中激发EMT和侵袭表型。29 TGF-β是在癌细胞中这种表型具有可塑性的强大诱导剂,它可以由癌症细胞自身或TME中的宿主细胞分泌。219例如,CAF-相关TGF-β信号转导增强体内外条件下癌症细胞的侵袭。220,221此外,基质金属蛋白酶和组织蛋白酶的催化活性改变了癌症细胞的生物物理特性,例如,通过上皮细胞中E-cadherin的裂解和整合素的调节,使癌症细胞能够机械适应不同硬度的基质。156,159,222,223
虽然E-cadherin的丢失和表型可塑性有助于癌症的侵袭,但并非所有肿瘤在转移播散过程中都会发生EMT样的转换。224,225保留上皮特征的癌细胞侵袭的机制尚不清楚,但越来越多的证据表明,CAF通过重塑细胞外基质,在细胞外基质中产生物理轨迹,从而使癌细胞集体侵袭。CAF与癌症细胞之间通过E-cadherin/N-cadherin连接的异型粘附触发癌症细胞的机械转导反应,实现集体侵袭。228其他TME细胞也可以促进癌细胞侵袭(图2)。例如,神经周侵袭(PNI)是癌细胞沿着神经局部延伸的过程,在几种实体癌症类型中观察到,这与不良结果有关。229在PDAC相关PNI的小鼠模型中,PNI位点的施旺细胞释放CCL2,从而吸引炎症单核细胞。随后这些细胞分化为组织蛋白酶B产生的巨噬细胞,增强神经侵袭。230在小鼠乳腺癌症模型中进行的活体显微镜(IVM)研究表明,EGFR+癌细胞的侵袭和迁移依赖于产生EGF的TAMs的结合。96,231总之,这些发现表明CAF、免疫细胞和组织-受体细胞的协同作用促进了癌症细胞的侵袭行为。
在健康组织中,基底膜在上皮细胞和下层基质之间形成物理屏障。232必须突破此屏障,才能使癌症细胞侵入周围组织(图1)。癌症细胞做到这一点的能力取决于多种因素的组合,包括它们的内部编程、ECM的结构和来自TME212,233的信号物质。CAF是通过分泌蛋白酶重塑基底膜和ECM网络起到关键作用,同时也通过施加收缩力在基底膜中产生间隙,从而给予癌细胞穿梭机会。234,235癌细胞随后受到ECM的组成和机械特性以及间质流体压力的影响,这影响了它们的迁徙和入侵能力。217,236它们可以通过整合素和其他跨膜受体感知重塑和交联的ECM分子,从而影响癌症细胞内在信号传递并增强侵袭和迁移。217,237例如,坚硬的ECM触发癌症细胞上的整合素聚集,从而刺激FAK/Src复合物的组装和PI3K/Akt和ERK信号的下游激活,促进癌症细胞的侵袭、迁移和生存。217在各种小鼠肿瘤模型中,药物或基因抑制ECM重塑和ECM交联,抑制FAK,以及其他减少基质硬化或癌症细胞对僵硬ECM减弱肿瘤发生的反应的方法。152,217,223,238
癌细胞渗入血管
转移的串联中下一步是速率控制,将癌细胞注入血液或淋巴循环(图1)。癌症细胞跨越内皮层进入循环的机制是复杂的,依赖于环境,并受癌症细胞固有特征、ECM的物理特性和血管系统类型、微环境物质以及缺氧程度的影响239(图2)。如上所述,肿瘤中的血管系统经常受损。血管基底膜和内皮屏障可能被破坏,从而增加血管渗出并促进癌症细胞的灌注。180小鼠IVM研究为灌注过程提供了关键见解。240 TAM通常与癌症细胞的灌注有关。196,240–244在植入PyMT乳腺肿瘤的小鼠体内进行的IVM和机械实验表明,位于血管近端的CXCL12分泌CAF可以将TAM和伴随的癌症细胞吸引到血管周围区域,在血管周围区域进行静脉注射。245 TIE2+血管周围TAM诱导的VEGF-A信号传导导致血管连接的局部丢失,导致血管通透性的短暂增加,从而促进癌症细胞的灌注。196因此,巨噬细胞耗竭可降低血管通透性和循环肿瘤细胞(CTC)的数量。196除了创建进入血管系统的通道外,TAM还直接对癌症细胞进行重新编程,以进行灌注过程。巨噬细胞激活癌症细胞中的RhoA信号,从而诱导癌症细胞侵入体的形成和随后在玻璃体中的灌注。244此外,TAM通过Notch-Jagged信号促进癌症干细胞的编程,导致缓慢、富含侵入体的癌症细胞表型增强灌注。242,243含有表达VEGF的TIE2+巨噬细胞、癌症细胞和内皮细胞的三重结构,也称为转移性肿瘤微环境(TME of metastasis, TMEM)的“通道”,以接触依赖的方式促进灌注(图2)。已在人类乳腺肿瘤中观察到TMEM,其密度预测远处转移的风险增加。246,247乳腺癌症患者和小鼠乳腺癌症模型的新辅助化疗通过促进Tie2+/VEGFhi巨噬细胞向肿瘤的动员,增加了TMEM位点的密度和活性,这与化疗治疗的实验模型中CTC和转移灶的增加有关。248TIE2抑制性逆转了化疗介导的前介导反应。248 TMEM门在静脉注射中的相关性主要来源于对乳腺癌症的研究。在其他类型的癌症中,是否有类似的机制是静脉滴注的基础尚待确定。此外,除了巨噬细胞和内皮细胞外,中性粒细胞、周细胞、CAF、脂肪细胞和TME的机械特征,包括ECM结构和间质流体压力,也通过直接或间接机制影响癌症细胞的灌注。239淋巴管内灌注是癌症细胞传播的另一种途径,尽管其潜在机制尚不完全清楚。肿瘤内淋巴管经常受压,淋巴管的结构与血管的结构不同,因此可能需要不同的浸润模式。206,249淋巴路径对远处转移形式的重要性存在争议,可能取决于器官。249
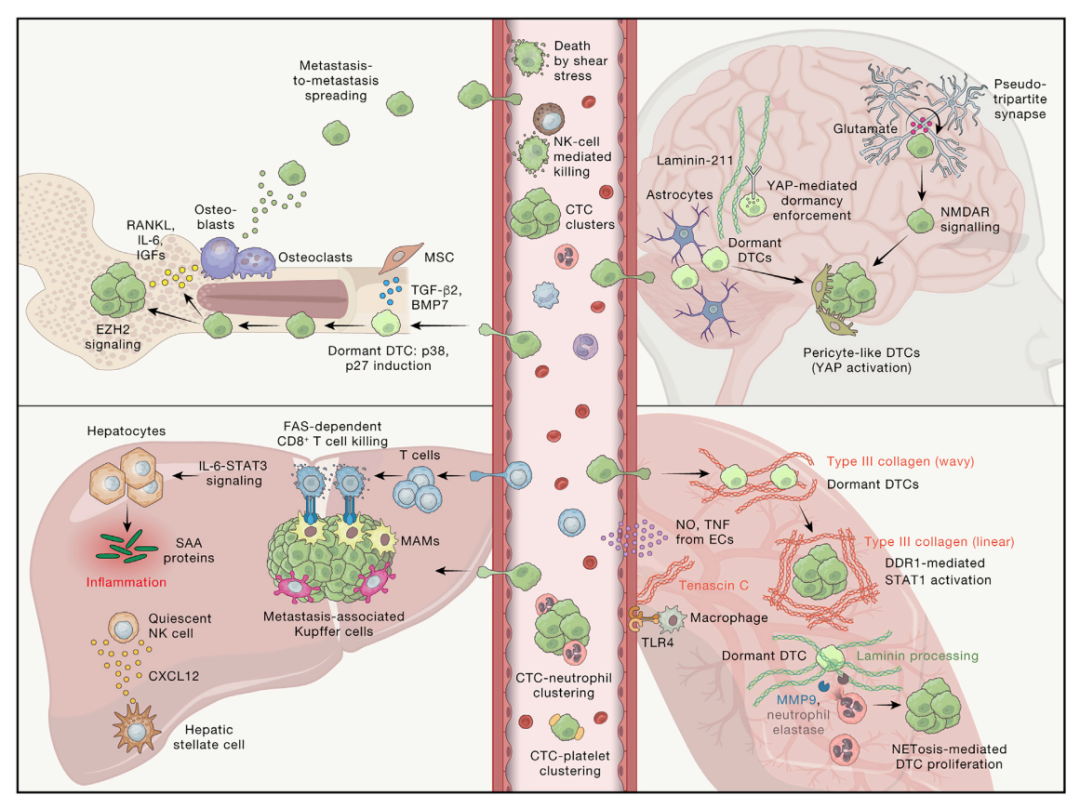
图3. 转移细胞在不同组织环境中命运
原发性肿瘤的远处转移——转移前生态环境的形成
发展中的肿瘤对宿主的影响并不局限于局部TME(图1)。通过旁分泌效应,原发肿瘤触发一连串事件,在远处器官中产生癌症细胞传导微环境,然后发生转移扩散。250我们了解到原发肿瘤通过为未来播散的远处位置做好准备,即转移前龛位,其所到位置远超其边界,导致我们对转移的理解发生了范式转变。最初在使用LLC肺和B16黑色素瘤肿瘤模型的研究中报道了转移前生存环境的存在。250研究表明,这些原发性肿瘤触发了VEGF和PIGF介导的MMP9在远处肺EC和巨噬细胞中的诱导,从而促进了肺转移的形成。MMP9在癌症患者的肺ECs中也有所上调,原发肿瘤位于肺部以外的其他器官,这在非癌症患者中未观察到。251另一项使用相同小鼠肿瘤模型的研究报告称,纤连蛋白(一种VLA-4配体)在转移前远处器官的成纤维细胞中被诱导,这指导了前转移VEGFR1+VLA-4+BM衍生的造血原始细胞的积累。重要的是,在癌症患者的常见转移部位也观察到VEFGR1+细胞簇,但在非癌症患者中未观察到。252自从这项开创性的研究以来,我们在分子和细胞机制方面的知识取得了实质性进展,这些分子和细胞机理是前转移微环境的基础,是扩散癌症细胞的肥沃土壤。250,253触发一系列系统性变化导致转移前微环境生成的起始信号包括肿瘤衍生的可溶性介质,最显著的是G-CSF、VEGF-A、PLGF、TGFβ、S100蛋白和TNF,以及携带肿瘤物质的EVs,这些肿瘤物质可以转移到远处器官中的BM细胞和固有细胞 。250其中一些介质影响BM微环境,在那里它们激活并编程免疫细胞及其祖细胞,以动员到未来的转移位点。10其他肿瘤分泌介质直接修饰远处器官。例如,低氧4T1乳腺癌症细胞分泌的LOX通过诱导破骨细胞发生而破坏了正常的骨内稳态,从而促进了CTCs的归巢和定植。254在LLC和B16荷瘤小鼠中,负载小核RNA的肿瘤衍生EVs激活肺上皮细胞中的Toll样受体3(TLR3),刺激中性粒细胞化学吸引介质的释放,通过中性粒细胞募集最终形成肺转移前微环境。255这些和其他最近的研究表明,原发性肿瘤破坏了远处器官中不同组织固有细胞和新动员的骨髓衍生免疫细胞之间的串扰,从而有助于转移前的微环境形成。250,253,256组织固有细胞的重要参与是转移前微环境形成中器官特异性差异的基础,部分解释了转移的器官趋向性。另一个有助于转移扩散器官特异性的因素是由肿瘤EV 粘附分子的表达决定的。在小鼠模型中,观察到根据整合素表达谱,肿瘤衍生的 EV 归巢到不同的远处器官,并且根据器官,不同的固有细胞表现出对肿瘤 EV 的摄取。因此,接种肿瘤分泌的 EV 可以改变癌细胞的器官趋化行为。 257此外,在癌症患者中,发现了具有特定整合素表达模式的EV,这些EV与转移的位置相关。257
原发性肿瘤使宿主为转移性疾病做好准备的另一个机制是通过诱导肿瘤诱导的全身炎症和免疫抑制,有利于扩散的癌症细胞的免疫逃逸。258例如,原发性Trp53缺陷型小鼠乳腺肿瘤中分泌IL1b的TAMs诱导IL-17和G-CSF依赖性免疫抑制中性粒细胞从骨髓细胞到远处器官,通过抑制CD8+T细胞促进转移扩散到肺部和淋巴结。11,12,81,259原发性肿瘤诱导的全身免疫抑制不仅影响未来的转移部位,而且影响整个宿主,因此在形式上不属于转移前微环境形成的概念。然而,系统动员的免疫抑制性骨髓细胞可能会触发组织环境特异性程序,以实现器官特异性转移。例如,在乳腺癌小鼠模型中,全身性调动分泌 IL-1β 的中性粒细胞可增强肺部外膜成纤维细胞分泌前列腺素 E3,从而导致抗肿瘤免疫力降低和肺转移增强。260
综上,虽然确切的旁分泌介质、细胞参与者和转移前微环境形成的级联事件可能因肿瘤类型而异,但在远处器官中产生的许可微环境的关键特征包括血管通透性增加、ECM 重塑、固有细胞的改变(包括成纤维细胞和上皮细胞)、BM 衍生细胞的动员和免疫抑制。下文将进一步探讨不同器官微环境对转移细胞命运的影响。
CTC 与循环中的生存之战
经过通常漫长的进化和适应过程,逐渐在原发部位塑造TME,一旦肿瘤细胞侵入循环(血液或淋巴),它们就会在这种外来微环境中立即受到一系列不同的袭击和挑战(图3)。在更常见的血道扩散情况下,包括细胞脱落引起的失巢凋亡;血液循环中的高切力和免疫介导的攻击,共同导致大多数 CTC 死亡。根据临床和前临床分析,据估计,每天每克组织中有 20,000 到 700,000 个 CTC 从实体瘤中脱落,具体取决于所分析的肿瘤类型。使用多种基因工程小鼠模型 (GEMM) 进行的详细血液交换分析估计,循环中内源性、自然脱落的 CTC 的半衰期为几分钟。261 有趣的是,最近的一项研究还发现其存在到 CTC 释放的昼夜节律性。262,263 然而,绝大多数 CTC 会死亡,这突显了传播过程的高低效性质,这是侵袭-转移级联中的关键限速步骤之一(图 1)。264,265
尽管如此,对于一小部分在循环中存活下来的 CTC,它们可以通过多种机制逃避破坏。这些包括 CTC 聚类,它通过诱导 NANOG、SOX2 和 OCT4266 来促进韧性,其与特定免疫细胞如中性粒细胞或血小板相关;相反,也逃避其他类型的细胞毒性免疫细胞(包括 NK 细胞)的作用(图 3)。事实上,CTC 与中性粒细胞的聚集以 VCAM-1 依赖性方式导致循环中 CTC 增殖增加,从而促进更有效的转移定植。 267 与这些机制见解一致,CTC 簇与单个 CTC 的富集通常患者预后较差相关,266 在循环中中性粒细胞与淋巴细胞的高比例与多种癌症的不良预后相关。 268
血液中另一种高度丰富的细胞类型是血小板。其长期以来被认为是 CTC 存活的关键促进剂,通过多种机制,包括增强 CTC 粘附和聚集,导致在 CTC 周围形成 “血小板斗篷”帮助它们逃避来自物理压力和免疫系统的监视269(图 3)。一种逃避免疫攻击的机制涉及一种分子模拟,其中含有MHC I类囊泡从血小板转移到肿瘤细胞表面保护CTC使其不被NK细胞识别。270相反,CTC可以激活血小板,例如通过G蛋白偶联受体(GPCR)CD97。271这导致通过CD97-LPAR信号传导增强的肿瘤细胞侵袭和促进血管舒张的ATP释放,从而导致CTC从循环中外渗,如以下节所述。
对抗中性粒细胞和血小板的CTC保护作用是NK细胞、细胞毒性T细胞、DC和其他免疫监测的破坏力。鉴于这些多样而快速的细胞相互作用发生在快速流动的循环中,这些相互作用是否只是以随机的方式发生,取决于哪种类型的免疫细胞首先与CTC相互作用,或者是否存在最终决定CTC命运的动态免疫“战斗”,这仍然是一个悬而未决的问题。诉于患者通过体液检查CTC可以作为一种微创手段来跟踪疾病发展,包括治疗反应和适应性耐药性的出现,并可能有助于回答这个问题。这些分析揭示了除了CTC和各种免疫细胞外,还有许多因素,包括EVs和非编码RNA,它们也可能影响CTC的生存能力,从而具有预后相关性。272然而,事实证明,检测和分离CTC是非常具有挑战性。从干预角度来看,这类分析当然是最有价值的。
器官趋向性和外渗
对于在循环中存活下来的一小部分 CTC,其转移过程中的下一个限速步骤是外渗到下级器官中。这在一定程度上是由每种原发性癌症的潜在器官趋向性决定的,被称为佩吉特在 1880 年代首次创造的“种子和土壤”假说。273 转移趋向性可能非常局限;例如,乳腺癌主要扩散到肺、肝、骨和脑,而前列腺癌则极有可能扩散到骨骼。 274这种趋向器官性受多种机制的影响,包括趋化因子、代谢物和 EV 等因素发出的信号,这些因素有助于 CTC 定向迁移到特定器官。此外,CTC 采取的特定循环途径以及癌细胞为进入特定器官必须跨越的不同血管屏障进一步影响了它们的最终目的地(在 Massague 和 Ganesh 中回顾265)。这种血管多样性通过比较 CTC 穿过大部分有孔的毛细血管进入 BM 的相对容易程度与穿越血脑屏障的多个紧密结合的细胞层的巨大挑战得到例证。 275,276
对于外渗过程,肿瘤细胞必须先停滞并附着于内皮腔,同时不断受到周围快速流动血流的高切力影响(图 3)。该步骤由肿瘤细胞和 ECs 表达的细胞粘附分子及其配体、整合素和 ECM 成分促进 277且与血液白细胞滚动、粘附和外渗所涉及的分子机制有一些相似之处。 278血小板和中性粒细胞可能仍与 CTC 一起移动,它们可以分别通过选择素或 GPCR 或通过产生中性粒细胞胞外陷阱 (NETs) 进一步增强肿瘤细胞与脉管系统的粘附。271,279
粘附后,CTC 接下来穿过 EC 连接处,也可能穿过额外的血管细胞层(例如,周细胞、平滑肌细胞)和 ECM,以进入新器官实质组织(图 3)。这通常需要细胞粘附分子(包括连接粘附分子、钙粘蛋白等)水解和、或降解,特别是多细胞 CTC 簇穿过脉管系统。癌细胞不仅依赖于它们为此步骤产生的蛋白酶和降解酶,280,281 它们还可以通过产生 NET 触发非癌细胞(包括血小板、单核细胞和中性粒细胞)释放上述酶。鉴于免疫细胞善于通过身体的不同器官来执行其生理功能,因此癌细胞可能会经历一种“免疫拟态”,从而产生通常富含免疫细胞的因子,包括趋化因子、蛋白酶和细胞粘附分子。280,281此外,如肺部 IVM 所示,未来转移部位的固有免疫细胞可进一步促进 CTC 通过门道外渗,282 与原发部位的内渗非常相似,如上文所述。在大脑中也揭示了常驻小胶质细胞对转移细胞外渗的类似增强283(图 3)。
因此,表型和物理水平的肿瘤细胞可塑性是赋予生存所需适应性的关键特征。 284 事实上,CTC 还可以通过非蛋白水解机制渗出,如滞胀,这涉及机械变形,一次挤压一个细胞通过EC连接。 285这种外渗模式对于从淋巴管进入淋巴结的CTC来说也更为典型。虽然已经报道了几种癌症的淋巴传播、神经周围迁移和胸膜腔或其他身体空间的生长,265但CTC运输的这些其他途径远低于血道扩散。最近的一项研究发现,淋巴结的转移定殖本身并不是转移克隆随后进化的枢纽,而是导致免疫抑制Tregs286介导的系统性肿瘤特异性免疫耐受。这使得远处组织更适合通过血液循环进行转移接种。外渗后,CTC通常留在血管附近,这对决定其命运至关重要,下一节将对此进行讨论。
肿瘤转移扩散以及肿瘤休眠和生长之间的复杂相互作用
播散性肿瘤细胞(DTC)在外渗到第二个部位后,面临来自外来组织环境的一系列新挑战,并且绝大多数肿瘤细胞再次被宿主防御机制杀死,包括免疫监测,表明它们接收来自血管的调节提示。事实上,来自PVN的分子信号,以及组织特异性微环境,如骨骼中的内皮微环境,最初似乎将DTC保持在休眠状态,这可能会保护它们免受免疫系统的识别和杀死(图3)。休眠代表转移级联中最不为人所知的阶段,部分原因是研究这些稀有细胞的固有挑战,这些细胞停止增殖并可以在静止状态下存活,有时长达数年至数十年。然而,最近包括那些利用活体成像能力的研究,240,282,288 开始揭示控制休眠启动、休眠在潜伏期的维持、休眠重新出现的机制的重要见解,尤其是与微环境的关键相互作用这种对器官特异性转移的精细调节。
骨转移
骨骼是与 DTC 生物学相关的研究最多的器官之一,部分原因是大部分患者发现该部位有微转移,尤其是乳腺癌和前列腺癌。289 骨骼微环境是多种不同细胞的家园,包括组织固有成骨细胞、破骨细胞和骨细胞,以及脂肪细胞、丰富的血管和免疫细胞,以及丰富的骨髓和 ECM289(图 3)。总之,这导致了一种动态的相互作用,在稳态下调节造血干细胞(HSC)的发育,但最终可能会参与骨转移伴随骨破坏和骨折的“恶性循环”。
在 DTC 定植的最早阶段,这些细胞可以占据骨骼中的不同微环境,大部分 DTC 位于骨髓,通常早在诊断为明显转移之前。289,290 例如,在 HSC 微环境中,NG2+/Nestin+ MSCs 产生 TGF-β2 和 BMP7,通过 p38 激酶信号和 p27 诱导激活 DTC 中的静止通路。 291在雌激素受体(ER)+乳腺癌患者中,无全身复发证据的患者骨髓血浆中 TGF-β2 和 BMP7 水平较高骨髓血浆中TGF-β2和BMP7水平较高。因此,TGF-β2的MSC缺失或MSC特异性缺失导致小鼠休眠骨DTC的转移生长。291骨内膜微环境还促进了 DTC 和骨固有细胞之间的双向相互作用,这可能最终促进疾病进展。癌细胞可诱导成骨细胞和破骨细胞分泌包括 RANKL、IL-6、IGF 和基质降解酶在内的因子,这些因子共同促进转移生长、骨溶解和骨骼变化,这些都是晚期骨转移的许多临床表现的基础。290
结合体内系统发育条码追踪的前临床实验表明,骨微环境中成骨细胞衍生的细胞因子可以通过 EZH2 依赖性方式通过表观遗传调节促进乳腺或前列腺 DTC 的干性。292,293 这增强了它们向其他器官的进一步传播,称为多器官-转移-转移-扩散,这可以通过小鼠中的 EZH2 抑制作用显着降低。 292这些结果也与临床观察结果一致,即骨骼通常是首先检测到乳腺癌和前列腺癌转移的部位。289,290 此外,骨骼微环境可以表观遗传调节 DTC 中的 ER 表达,从而使细胞对内分泌治疗产生耐药性,293 建模则是临床中的一个主要挑战。同样,在患者中经常会遇到 DTC 对全身化疗的耐药性。这归因于静止的 DTC,它们不会增殖,因此不会成为此类疗法的目标,并且可能会在以后从休眠状态中出现。然而,近期前临床研究发现,骨 PVN 可以通过血管粘附分子包括整合素 β1、αvβ3 和 VCAM-1294保护 DTC 免受化疗,与其细胞周期状态无关。因此,阻断 PVN 和 DTC 之间整合素介导的相互作用可使这些细胞对化疗敏感并显著提高存活率。294
肺转移
在肺部的DTC 的播种、休眠和生长也进行了研究,发现肺部是转移性扩散的主要部位。事实上,肺部广泛的血管化和较大的表面积对正常的肺功能至关重要,CTC 有很多机会侵入、渗出和定植该器官(图 3)。通过 IVM 揭示了这一过程的关键见解,表明肿瘤细胞已经在原发部位获得了由转录调节因子 NR2F1 控制的促扩散和休眠表型,并且在巨噬细胞附近进一步丰富。 282 这种状态最初维持在 DTC 到达肺部后的生长过程中丢失。 282与其他器官相似,来自 PVN 的物质对于决定 DTC 命运至关重要,包括 III 型胶原蛋白和生腱蛋白 C 在内的 ECM 分子起着重要作用。295,296 成像分析结合二次谐波生成来评估胶原纤维方向,揭示了孤立的休眠 DTC 被非线性波浪方向的 III 型胶原蛋白包围。相比之下,在微转移的增殖诱导的同时,胶原组织和相关ECM重塑也发生了变化。295因此,通过靶向已鉴定的COL3A1-DDR1-STAT1途径来操纵这些不同的ECM微环境,被认为是维持DTC休眠的一种方法。295另一种调节肺部 DTC的分泌型 ECM 分子是生腱蛋白 C,它通过 TLR4 信号依次激活邻近的间质巨噬细胞,诱导 EC 分泌一氧化氮 (NO) 和 TNF,从而产生促转移性 PVN 微环境。 296
有趣的是,间质调节肺 DTC 休眠的重要性在衰老背景下的前临床研究中也很明显。297 虽然衰老皮肤中的真皮成纤维细胞抑制黑色素瘤细胞的生长,但它们可以通过可溶性 WNT 、拮抗剂 sFRP2驱动黑色素瘤表型转换和传播。298当这些 DTC 到达肺部时,它们会遇到相关的 sFRP1 拮抗剂,这种拮抗剂由衰老的肺成纤维细胞以更高的水平分泌。这导致黑色素瘤细胞中 WNT5A 以 PROS1-AXL 依赖性方式受到抑制,从而克服休眠并最终导致随后的黑色素瘤生长。 297因此,对该旁分泌信号通路不同成分的基因操作导致肺转移发生改变,297 这可能对针对特定老年人群的癌症进行治疗靶向具有重要意义。299 另一种可以唤醒休眠癌细胞的诱因是炎症。事实上,持续的炎症,例如接触烟草的烟雾,会诱发 NETs 的形成。83 NETs 充满了蛋白酶,包括 MMP9 和中性粒细胞弹性蛋白酶。这些蛋白酶的释放重塑了肺中的层粘连蛋白,导致休眠癌细胞以整合素 a3b1 依赖性方式增殖 83(图 3)。
肝转移
肝脏是一个也是常见的转移器官,部分原因是它广泛血管化,具有来自肝门静脉和肝动脉的双重血液供应,还因为肝血管系统高度联通,从而促进 CTC 外渗 300(图 3)。与上述器官一样,基质和 ECM 的改变对于调节肝脏中的转移生长也很重要。30 0 例如,在一项针对结直肠癌患者的大型研究中,肝纤维化评分高的患者在肝转移和复发方面的结果明显更差。301 此外,组织固有肝细胞还被证明通过形成促转移的基质在前临床模型中促进肝转移。302在胰腺肿瘤发生的早期,肝脏中的肝细胞通过激活IL-6-STAT3信号传导和随后增加血清淀粉样蛋白A(SAA)蛋白的产生来协调炎症反应。对肝转移患者的分析揭示了类似变化。302这导致肝脏中纤维化和免疫微环境的改变,这与增强小鼠的转移接种有关。有趣的是,通过肝细胞特异性缺失去除SAA蛋白或Stat3,阻断了促转移的微环境。302
另一个肝脏固有细胞群,即肝星状细胞,也在调节乳腺癌症细胞在该器官中的命运方面发挥着重要作用。303星状细胞可以通过分泌免疫抑制性CXCL12趋化因子来驱动纤维化损伤,使NK细胞静止。这抑制了NK细胞的关键免疫监视功能,导致DTC从休眠中重新出现。303同样有意思的研究发现,肝转移的存在会通过从系统循环中抽取激活的CD8+T细胞,对免疫疗法的疗效产生负面影响。304发现与肝转移中巨噬细胞的相互作用以 Fas 依赖性方式驱动 T 细胞杀伤。这种影响在前临床模型中通过肝脏定向放疗被克服——消除了这些免疫抑制性巨噬细胞,从而减少了 T 细胞抽取。鉴于还发现肝转移患者的 T 细胞数量、多样性和功能减少,这些结果可能对提高该患者免疫治疗效果的策略具有重要意义。 304
脑转移
在大脑中,组织固有星形胶质细胞构成血脑屏障 (the blood-brain barrier, BBB) 的重要组成部分,并且已发现静止的 DTC 位于被星形胶质细胞末端包围的 PVN 中 288 (图 3)。星形胶质细胞衍生的基底膜蛋白 laminin-211 可以通过诱导肌营养不良聚糖受体与 DTC 中的 YAP 结合来加强这种休眠。这可以防止 YAP 运输到细胞核,从而阻止其促转移功能。相反,增殖的 DTC 仅与被剥离星形胶质细胞及其末端足的血管结构相关。因此,这些不同分子成分的体内调节导致脑转移生长。 288 YAP 激活也与 DTC 自身获得周细胞样特征有关,这通过细胞粘附分子 L1CAM 促进它们在大脑和其他转移部位的伸长和转移生长。 305在 DTC 可塑性的另一个例子中,这些细胞甚至可以将自身整合到大脑的神经网络中。 306 在这种情况下,DTC 将自身定位在两个谷氨酸能神经元之间的现有突触附近,形成“伪三联体”突触。这导致神经元释放谷氨酸,从而导致 N-甲基-D-天冬氨酸受体 (NMDAR) 信号传导和随后的转移生长。 306 此外NMDAR 通路还可以促进胰腺神经内分泌肿瘤的侵袭,307 而新兴癌症神经科学领域 308 代表了肿瘤对宿主微环境的一种特别神秘的合作。
总之,这些说明性的例子强调了不同器官微环境在调节DTC命运中的重要性,这些微环境由独特的组织固有细胞类型以及募集的免疫细胞组成。鉴于DTC可以潜伏数年至数十年,而其激活是一个重大的临床挑战,因此必须充分了解其潜在机制,因为这一关键阶段仍然是癌症领域的“黑匣子”。类似地,与原发性肿瘤相比,治疗休眠DTC本身就具有挑战性,TME的操作可能是成功的关键。
结束语:机遇与挑战
从TME研究领域一开始,针对TME中的细胞、过程和信号通路的治疗被视为一种有希望的方案,原则上可以推广到所有癌症类型。与基因组不稳定的癌症细胞相比,稳定的肿瘤细胞更容易靶向。此外,由于类似的原因,获得性耐药性的发展被认为不太可能,为其至少是通过针对癌细胞定向疗法观察到的基于突变的选择。最后,希望 TME 疗法甚至可以代表一种可以应用于任何肿瘤类型的通用方法,无论它在哪个器官中发展。
然而,随着近年来该领域在范围和理解上的发展,我们开始意识到这些早期预测过于简单化。正如这篇综述中所讨论的,我们现在认识到 TME 的巨大复杂性和相互关联性,以及它在不同器官和患者之间的多样性。我们还认识到,适应性和内在抵抗力可能成为 TME 定向疗法的障碍。此外,显而易见,包括化学疗法和放射疗法在内的标准护理疗法会引起 TME 的变化,从而以癌细胞外在的方式调节其治疗效果,从而增强或干扰反应。 309例如,放疗和某些化疗可以引发免疫原性细胞死亡,这会通过适应性免疫系统来增强它们的疗效。310 然而,在其他情况下,许多相同的治疗会引起炎症反应,包括通过 TAM从而干扰治疗反应 85,甚至可能推动转移扩散。 86
尽管着挑战,但正如上述,在针对TME的治疗策略的不断扩展方面也有很大的希望。1其中包括耗尽或“重新编程”TME中促癌宿主细胞的治疗、改变ECM、基质体和 EV 的干预措施、细胞疗法和疫苗、免疫检查点抑制剂。1现在的关键问题是如何以合理且最佳的方式组合这些不同的方法。事实上,在临床中,开放联合试验的数量远大于符合条件的患者人数。311 这是一个巨大的障碍。使用可靠的临床前模型提供了一种首先系统地评估所有逻辑组合的方法。事实上,最近对免疫疗法临床试验的讨论分析强调了在新疗法评估中纳入准确的临床前的必要性,该分析发现,超过70% 的此类疗法在试验中没有先前重要的临床前证据支持正在评估的组合。312 此外,大多数试验是在没有选择任何生物标志物的人群中进行的。 312这凸显了另一个难点:如何针对 TME 疗法对患者进行最佳分层,特别是对于不能直接为此目的进行活检的癌症。重要的是要确定循环免疫细胞或 EV 的相对浓度和/或表型分析是否可以代表肿瘤分析的替代方法。 313,314再有关键问题是如何靶向已经扩散并进入休眠状态的癌细胞,因为调节后从休眠状态出现无疑涉及 TME 的调节(图 3)。
最后,我们将简要强调一些最近的例子,这些例子可以为如何针对 TME 进行治疗提供“路线图”,包括致力于将免疫疗法的疗效从目前的癌症患者亚群扩展到更广泛的患者群体。事实上,越来越多的证据表明 TME 在调节免疫治疗效果(如化学疗法和放射疗法)方面也起着至关重要的作用,正如本综述所强调的那样。这可能不同于细胞毒性 T 细胞的物理排斥,导致免疫排斥 TME,也可能产生免疫抑制的 TME,其中 T 细胞存在但由于与免疫抑制细胞的相互作用而功能失调。 40因此,将 ICB 或基于细胞的免疫疗法与 TME 调节相结合的策略正在临床前模型中积极探索或已经在进行临床评估。 315,316 其中包括施加于免疫刺激细胞因子,如 IL-12 和 IL-15 ,激活 NK 细胞317; 通过工程化的 MSC318 将 IL-2(T 细胞的关键细胞因子)局部递送至 TME;以及 IL-2 变体的开发,目的提高 T 细胞浓度和细胞毒性功能,同时避免对免疫抑制性 Treg 产生类似影响。 316
其他组合利用抗血管生成抑制剂,这是首批 TME 靶向治疗之一,随后被发现具有强大的血管调节作用。 319例如,抗 VEGF 疗法可以促进免疫刺激细胞的浸润,阻断 TME 中的免疫抑制作用,并改善药物作用于靶点。320 血管系统和免疫系统之间的复杂相互作用还可以通过干预来增强高内皮小静脉(high endothelial venules, HEV)的形成,因为肿瘤相关的HEV、淋巴细胞浸润增加和某些癌症的良好预后之间存在关联。321这些和其他血管靶向疗法现在被广泛纳入与 ICB 和细胞疗法的组合策略中,以增强肿瘤中的免疫细胞浸润和细胞毒性。1,320,321 如最近的代表性研究所揭示的,以新的组合或通过将“冷”转化为“热”TME的不同给药方案重新调整临床批准的治疗方法的用途,代表了将临床前模型的研究结果快速转化为临床的另一种策略。 1,322,323
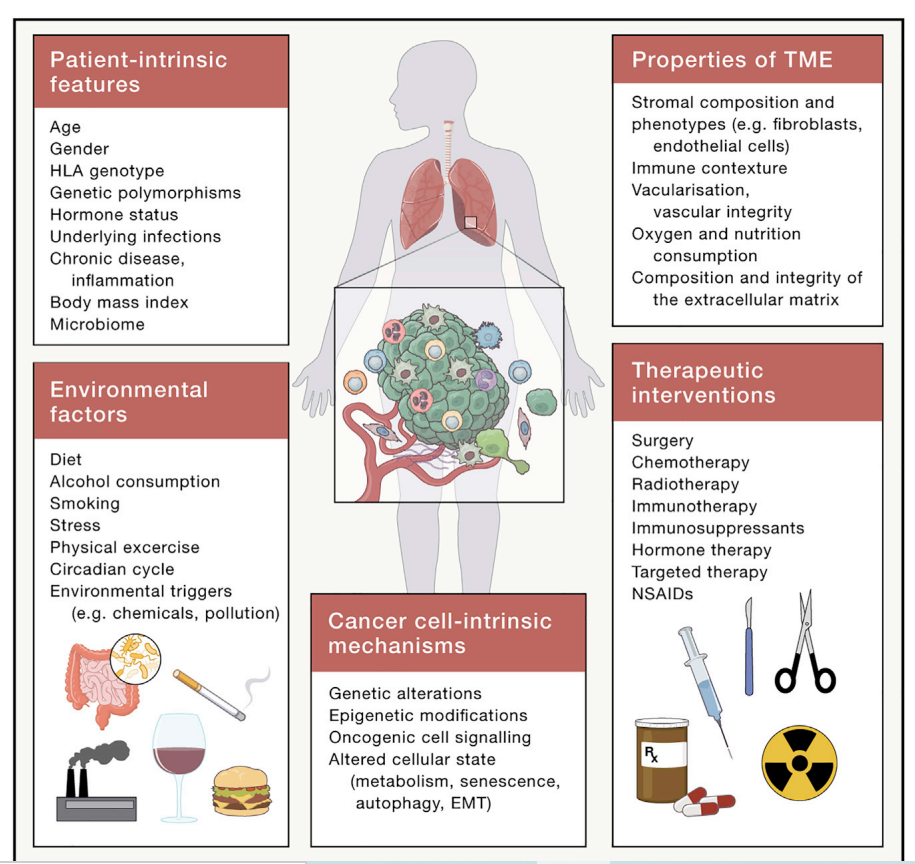
图4. 多种内因和环境因素影响患者和肿瘤微环境
展望未来,我们乐观地认为,将在未来几年取得关键进展,以充分实现针对TME的承诺。与其一次研究一种细胞类型的TME,我们希望该领域采用全面的系统级方法,分析和整合TME的所有复杂性,以识别和治疗关键节点。整合多模态数据和先进的计算分析,包括使用人工智能,324,325对实现这一目标极有帮助。我们还预想生物工程将取得重大进展,这将使大规模测试的平台成为可能,例如准确概括器官特异性TMEs的离体类器官和组织切片。326,327了解肿瘤本身之外的额外复杂性层,包括系统影响和外部环境,对该领域也至关重要(图4)。例如,微生物、饮食、运动和代谢如何影响TME和治疗反应?此外,个体患者的潜在生理学有什么贡献,如肥胖、恶病质、昼夜节律、炎症和衰老。最后,我们可以期待外部环境(如污染和致癌物暴露)对炎症和TME的影响有新的观点,这应该会激发迫切需要的公共卫生响应。总之,通过整合和利用这些关键观点,我们乐观地认为,在不久的将来,我们将能够以TME为治疗目标,造福更多患者。
参考文献(略)
参考资料:de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023 Mar 13;41(3):374-403. doi: 10.1016/j.ccell.2023.02.016. PMID: 36917948.
版权声明:本网站所有注明来源“医微客”的文字、图片和音视频资料,版权均属于医微客所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源:”医微客”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,转载仅作观点分享,版权归原作者所有。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。 本站拥有对此声明的最终解释权。































 关注公众号
关注公众号 安卓客户端
安卓客户端










发表评论
注册或登后即可发表评论
登录注册
全部评论(0)